| Issue |
A&A
Volume 620, December 2018
|
|
|---|---|---|
| Article Number | A170 | |
| Number of page(s) | 41 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201731667 | |
| Published online | 14 December 2018 | |
The ALMA-PILS survey: isotopic composition of oxygen-containing complex organic molecules toward IRAS 16293–2422B
1
Centre for Star and Planet Formation, Niels Bohr Institute & Natural History Museum of Denmark, University of Copenhagen,
Øster Voldgade 5–7,
1350
Copenhagen K.,
Denmark
e-mail: jeskj@nbi.ku.dk
2
I. Physikalisches Institut, Universität zu Köln,
Zülpicher Str. 77,
50937
Köln,
Germany
3
Laboratoire d’astrophysique de Bordeaux, Université de Bordeaux, CNRS,
B18N, allée Geoffroy Saint-Hilaire,
33615
Pessac,
France
4
Center for Space and Habitability (CSH), University of Bern,
Sidlerstrasse 5,
3012
Bern,
Switzerland
5
Harvard-Smithsonian Center for Astrophysics,
60 Garden Street,
Cambridge,
MA
02138,
USA
6
Department of Sea, Earth and Environment, Chalmers University of Technology, Onsala Space Observatory,
439 92
Onsala,
Sweden
7
INAF–Osservatorio Astrofisico di Arcetri,
Largo E. Fermi 5,
50125 Firenze,
Italy
8
Leiden Observatory, Leiden University,
PO Box 9513,
2300
RA Leiden,
The Netherlands
9
Max-Planck Institut für Extraterrestrische Physik (MPE),
Giessenbachstr. 1,
85748
Garching,
Germany
Received:
28
July
2017
Accepted:
27
August
2018
Context. One of the important questions of astrochemistry is how complex organic molecules, including potential prebiotic species, are formed in the envelopes around embedded protostars. The abundances of minor isotopologues of a molecule, in particular the D- and 13C-bearing variants, are sensitive to the densities, temperatures and timescales characteristic of the environment in which they form, and can therefore provide important constraints on the formation routes and conditions of individual species.
Aims. The aim of this paper is to systematically survey the deuteration and the 13C content of a variety of oxygen-bearing complex organic molecules on solar system scales toward the “B component” of the protostellar binary IRAS16293–2422.
Methods. We have used the data from an unbiased molecular line survey of the protostellar binary IRAS16293−2422 between 329 and 363 GHz from the Atacama Large Millimeter/submillimeter Array (ALMA). The data probe scales of 60 AU (diameter) where most of the organic molecules are expected to have sublimated off dust grains and be present in the gas phase. The deuterated and 13C isotopic species of ketene, acetaldehyde and formic acid, as well as deuterated ethanol, are detected unambiguously for the first time in the interstellar medium. These species are analysed together with the 13C isotopic species of ethanol, dimethyl ether and methyl formate along with mono-deuterated methanol, dimethyl ether and methyl formate.
Results. The complex organic molecules can be divided into two groups with one group, the simpler species, showing a D/H ratio of ≈2% and the other, the more complex species, D/H ratios of 4–8%. This division may reflect the formation time of each species in the ices before or during warm-up/infall of material through the protostellar envelope. No significant differences are seen in the deuteration of different functional groups for individual species, possibly a result of the short timescale for infall through the innermost warm regions where exchange reactions between different species may be taking place. The species show differences in excitation temperatures between 125 and 300 K. This likely reflects the binding energies of the individual species, in good agreement with what has previously been found for high-mass sources. For dimethyl ether, the 12C/13C ratio is found to be lower by up to a factor of 2 compared to typical ISM values similar to what has previously been inferred for glycolaldehyde. Tentative identifications suggest that the same may apply for 13C isotopologues of methyl formate and ethanol. If confirmed, this may be a clue to their formation at the late prestellar or early protostellar phases with an enhancement of the available 13C relative to 12C related to small differences in binding energies for CO isotopologues or the impact of FUV irradiation by the central protostar.
Conclusions. The results point to the importance of ice surface chemistry for the formation of these complex organic molecules at different stages in the evolution of embedded protostars and demonstrate the use of accurate isotope measurements for understanding the history of individual species.
Key words: astrochemistry / stars: formation / stars: protostars / ISM: molecules / ISM: individual objects: IRAS 16293-2422 / submillimeter: ISM
© ESO 2018
1 Introduction
The earliest stages of protostars are characterised by a rich chemistry, which in some cases leads to abundant complex organic, and even prebiotic, molecules present in the gas phase close to the young stars of both low and high mass. The general picture is that the formation of icy grain mantles and chemistry in and on grains facilitates the build-up of larger species through surface reactions starting from CO (e.g. Tielens & Hagen 1982; Hasegawa & Herbst 1993; Charnley 1997; Watanabe & Kouchi 2002; Fuchs et al. 2009; Öberg et al. 2009; Garrod 2013; Fedoseev et al. 2015; Chuang et al. 2017). However, a number of important open questions remain concerning the chemistry leading to the formation of these species, for example what specific reactions are dominant, what is the importance of UV irradiation, how important is the ice composition and the rate at which the ices are warming up.
The isotopic composition of the gas is a particular characteristic of the early stages of young stars, which may hold significant clues to some of these networks. It has for some time been recognised that the many species in the cold gas around embedded protostars are significantly enhanced in deuterium relative to hydrogen(e.g. van Dishoeck et al. 1995; Ceccarelli et al. 2001) compared to the cosmic D/H ratio of 1.5 −2.0 × 10−5 (e.g. Linsky 2003; Prodanović et al. 2010). In particular, the deuterated isotopologues of species such as formaldehyde (H2 CO) and methanol (CH3 OH) are found to be significantly enhanced upto levels of ~10% or above compared to the regular isotopologues in single-dish observations (e.g. Parise et al. 2006)and some of these sources even show detections of multiple deuterated species (e.g. CD2 HOH and CD3 OH; Parise et al. 2002, 2004) indicating very high degrees of deuteration (see also overview in Jørgensen et al. 2016).
These enhancements are thought to be a consequence of the exothermic reaction
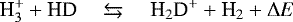 (1)
(1)
(where ΔE = 232 K), which followed by dissociative recombination of H2 D+ with electrons leads to an enhanced atomic D/H ratio that can be transferred to grains when the neutral species freeze-out (Tielens 1983). Other isotopic systems such as 12C- and 13C-containing isotopologues may also show variations compared to the local ISM due to the fractionation processes in the cold phases although the variations of those are expected to be at much smaller levels than those of deuterium vs. hydrogen. Isotope selective photodissociation or ion–molecule reactions in the gas through the reaction
 (2)
(2)
(where ΔE = 35 K; Langer et al. 1984; Furuya et al. 2011) may enhance the 13CO abundance in the gas phase relative to 12CO. Isotope-selective photodissociation may decrease the amount of 13 CO relative to 12CO, but at the same time enhancing the amount of 13C. Once on the grains, the enhanced 13CO or 13C can then be incorporated into the complex organic molecules. Other mechanisms, such as segregation between 12CO and 13CO in the ices due to slight differences in their binding energies (e.g. Smith et al. 2015) may also affect the amount of 12C relative to 13C that is available for incorporation into larger molecules.
Since all of these fractionation processes are very sensitive to the gas physics (density and temperature), it has therefore been suggested that measurements of the relative abundances of different isotopologues of individual molecules may hold strong clues to the formation pathways constrained by chemical models or alternatively to the physical conditions and early evolution of young stars through their pre- or protostellar stages (e.g. Charnley et al. 2004; Cazaux et al. 2011; Taquet et al. 2013, 2014). With the Atacama Large Millimeter/submillimeter Array (ALMA), it is becoming possible to push forward in providing accurate measurements of the isotopic composition of complex organics.
As an example of such a study, Belloche et al. (2016) presented a systematic survey of the deuteration of molecules toward the Sagittarius B2 high-mass star forming region in the central molecular zone close to the Galactic center. Utilising ALMA’s high sensitivity, Belloche et al. (2016) presented new detections as well as strict upper limits for the deuterated variants of a number of complex organics. Typically, the abundances of the deuterated species were found to be lower than what is seen in nearby molecular clouds and also predicted by astrochemical models. Belloche et al. (2016) speculated that this may reflect a lower overall deuterium abundance or the higher temperatures in the clouds toward the Galactic center.
One of the prime targets for similar studies of solar-type stars is the Class 0 protostar IRAS16293–2422. This source has long been recognised as having large abundances of deuterated species compared to high-mass protostellar sources (van Dishoeck et al. 1995)and was also the first solar-type protostar for which abundant complex organic molecules were found (Cazaux et al. 2003). Combining these two properties, it has been a natural target for deuteration studies. This is the source toward which the above-mentioned multi-deuterated species, as well as some deuterated complex organics, for example methyl formate (CH3 OCHO; Demyk et al. 2010) and dimethyl ether (CH3 OCH3, Richard et al. 2013), were first detected. Recent observations of the THz ground-state transitions of H2 D+ (Brünken et al. 2014) and D2 H+ (Harju et al. 2017) toward the cold envelope around IRAS16293–2422 suggest very similar amount of both cations in this source. Due to sensitivity limitations, however, the single-dish observations typically targeted the brightest, low excitation, transitions that in some cases may be optically thick both for the main isotopologues as well as for the deuterated variants. Furthermore, single-dish observations naturally encompass the entire protostellar envelope and are thus intrinsically biased toward emission on larger scales where most of the material is located and molecules may still be largely frozen-out on dust grains due to the lower temperatures.
Using ALMA we have recently completed a large unbiased survey, the Protostellar Interferometric Line Survey (PILS), of a key frequency window around 345 GHz (Jørgensen et al. 2016)1. These data are up to two orders of magnitude more sensitive than previous single-dish studies and thus provide excellent opportunities for systematic studies of complex organics and their isotopologues. In particular, the interferometric observations make it possible to zoom in on the inner 30–60 AU around the central protostars, i.e. solar system scales, where the temperatures are high and the ices have sublimated and the complex organic molecules therefore are all present in the gas phase. The initial results presented the first detections of the deuterated isotopologues of isocyanic acid and formamide (Coutens et al. 2016) as well as the deuterated and 13C-isotopologues of glycolaldehyde (Jørgensen et al. 2016). These species all show significant deuterium enhancements – although not on the levels hinted by the previous single-dish observations – but also indications of differences that may exactly reflect their chemistries. Specifically, glycolaldehyde is more enhanced in deuterium than the other species by a factor of a few. The 13C-isotopologues of glycolaldehyde further indicate an enhancement of the 13C isotopologues above that of the local ISM, again possibly reflecting formation of this species in the coldest part of the envelope.
In the present work, we present an extensive investigation into the deuteration and the 13C content of oxygen-bearing complex organic molecules. These include methanol, its next heavier homologue ethanol along with its isomer dimethyl ether, the dehydrogenated relatives of ethanol, acetaldehyde, and ketene, as well as methyl formate, an isomer of glycolaldehyde, and formic acid. The paper is laid out as follows. Section 2 summarises the main points about the ALMA data and spectroscopy while Sect. 3 describes the data analysis, including line identification and column density/abundance determinations of the complex organics and their isotopologues. Section 4 discusses the implications in relation to the formation of the species.
2 Data analysis
In the following, we present the results concerning identification of the isotopologues of oxygen-bearing complex organic molecules. Section 2.1 summarises the key aspects of the observations and Sect. 2.2 describes the information about laboratory spectroscopy. Further information about the history of detections and issues with the fits for individual species are given in Appendix A.
2.1 The ALMA PILS survey
In this paper we utilise data from the ALMA-PILS survey targeting the protostellar binary IRAS16293–2422 (IRAS16923 hereafter) using the Atacama Large Millimeter/submillimeter Array. An overview of the data and reduction as well as first results from the survey are presented in Jørgensen et al. (2016); here we highlight a few specifics. The PILS survey provides a full survey of IRAS16293 covering the frequency range in ALMA’s Band 7 from 329.1 to 362.9 GHz with 0.2 km s−1 spectral and ≈0.5′′ angular resolution (60 AU diameter) performed in ALMA’s Cycle 2 (project id: 2013.1.00278.S). Also, additional targeted observations were performed in ALMA’s Bands 3 (≈100 GHz) and 6 (≈ 230 GHz) taken as part of a Cycle 1 program (project-id: 2012.1.00712.S). The Band 7 part of the survey is one–two orders of magnitude more sensitive than previous surveys, reaching a sensitivity of ≈5 mJy beam−1 km s−1 across the entire frequency range. In this paper, we focus on the “B component” of the source, which, with its narrow lines (~1 km s−1; e.g. Jørgensen et al. 2011), is an ideal target for searches for rare species.
2.2 Laboratory spectroscopic information
The spectroscopic data were taken from the Cologne Database of Molecular Spectroscopy (CDMS; Müller et al. 2001, 2005) and the Jet Propulsion Laboratory (JPL; Pickett et al. 1998) catalogue. These databases compile, and in some cases extend, spectroscopic information and partition functions based on reports in literature. Some care must be taken when comparing across these spectroscopic entries. In particular, for some entries, the partition functions only contain the ground vibrational state while the excited vibrational states also may be populated at the temperatures of up to a few hundred Kelvin characterising the gas on small scales toward IRAS16293. Calculated column densities considering only the ground vibrational state will therefore underestimate the true values. We therefore carefully examined the original literature for each species, for example evaluating the vibrational correction factors, as described in the following. Particular attention was paid to make sure that the partition functions were in fact derived using the same degeneracies and thus line strengths as listed in the catalogs.
For the analysis of methanol, we used the 18O isotopologue using the catalogue entry from the CDMS. The entry is based on Fisher et al. (2007) who combined new far-infrared (FIR) data and rotational data with microwave accuracy summarised in Ikeda et al. (1998). The partition function should be converged at 300 K. For CH2DOH the entry from the JPL catalogue was used: this entry is based on Pearson et al. (2012). The partition function in that entry takes only the ground vibrational state into account. The vibrational correction factor is estimated to be 1.457 at 300 K based on torsional data from Lauvergnat et al. (2009). For CH3 OD transition frequencies from Duan et al. (2003) with published line strengths from Anderson et al. (1988) and with partition function values scaled with those from CH OH are used.
OH are used.
Ethanol, C2 H5OH, possesses two large amplitude motions, the torsions of the CH3 and the OHgroups. The methyl torsion is usually neglected in the ground vibrational state. The OH torsion leads to twodistinguishable conformers, the lower lying anti conformer and the doubly degenerate gauche conformer. Tunneling between the two gauche minima leads to two distinguishable states, the symmetric gauche+ and the antisymmetric gauche−. Pearson et al. (2008) present an extensive analysis of the ground-state rotational spectrum of ethanol and determined the energy differences with great accuracy. The results of the analysis are available in the JPL catalogue. Müller et al. (2016), however, found that the predicted intensities do not match those of ethanol emissions in an ALMA survey of Sagittarius (Sgr) B2(N2) at 3 mm. Intensity discrepancies were also detected in laboratory spectra (F. Lewen, unpublished) and in our ALMA data. A new catalogue entry was created for the CDMS as a consequence. The line and parameter files are those provided in the JPL catalogue. The entry is based on Pearson et al. (2008) with additional data in the range of our survey from Pearson et al. (1995, 1996) for anti and gauche ethanol, respectively.
Information on the 13C and D containing isotopomers of ethanol were taken from the CDMS. The entries were based on Bouchez et al. (2012) and Walters et al. (2015), respectively. The entries deal with the anti conformer only in each case which can be treated separately up to moderately high quantum numbers (Pearson et al. 1995). Transitions are modeled well for J + 2Ka ≤ 32 in the case of the 13C isotopomers, slightly higher in J or Ka for mono-deuterated ethanol. The column densities of ethanol with 13C or D have to be multiplied by ~2.69 in order to account for the presence of the gauche conformer. This factor was evaluated from the ground-state partition functions of the main isotopic species involving the anti conformer only and both conformers, respectively. The correction factors are expected to be very similar for the 13C isotopomers, but may be slightly larger for the deuterated isotopomers. The vibrational factor at 300 K is 2.824 (Durig et al. 1975).
The dimethyl ether, CH3 OCH3, data were taken from the CDMS. They are based on Endres et al. (2009) with additional data in the range of our survey from Groner et al. (1998). Data of mono-deuterated dimethyl ether were taken from Richard et al. (2013) and those of 13CH3OCH3 from Koerber et al. (2013). The partition function of the main isotopologue includes contributions from vibrational states up to v7 = 1 (Groner & Durig 1977; C. P. Endres, unpublished). The difference at 125 K is only a factor of 1.185.
For acetaldehyde, CH3CHO, the JPL catalogue entry based on Kleiner et al. (1996) is used. Experimental transition frequencies in the range of our survey are from Belov et al. (1993). Elkeurti et al. (2010) provide data on CH3 CDO while Margulès et al. (2015) published data on the isotopomers with one 13C. The partition function of the main isotopologue includes contributions from the first and second excited methyl torsional mode. Their contributions are 0.243 at 125 K for the first and 0.049 for the second.
The ketene entries are taken from the CDMS catalogue. The main, 13C and 18O data are based on Guarnieri & Huckauf (2003) with additional data in the range of our survey from Brown et al. (1990). The entries with one and two D are based on Guarnieri (2005). Vibrational contributions to the partition function are very small at 125 K.
We took the methyl formate (CH3OCHO) data from the JPL catalogue. The entry is based on Ilyushin et al. (2009) with data in the range of our survey from Plummer et al. (1984), Oesterling et al. (1999) and Maeda et al. (2008a). The CH3 OCDO data were based on Oesterling et al. (1995) and Margulès et al. (2009), and those of CH2DOCHO on Margulès et al., (2009; available in Coudert et al. 2013). We consulted the CDMS catalogue for CH3 O13CHO data. The entry is based on Carvajal et al. (2010) with additional data in the range of our survey from Willaert et al. (2006) and Maeda et al. (2008a,b). The vibrational factors for entries containing only vt = 0 and those containing both vt = 0 and vt = 1 are listed in the CDMS.
Formic acid, HCOOH, occurs in two conformers, trans and cis, with the latter being almost 2000 K higher in energy. Predictions for the main species were taken from the CDMS which were based on Winnewisser et al. (2002a). Predictions for H13COOH, DCOOH and HCOOD were taken from the JPL catalogue. They were based on Lattanzi et al. (2008) with (additional) data in the range of our survey Winnewisser et al. (2002b); Baskakov et al. (2006) for H13COOH, Baskakov (1996) for DCOOH and summarised in Baskakov et al. (1999) for HCOOD. Formic acid has two relatively low-lying vibrational modes, ν7 and ν9. Vibrational corrections to the ground state partition function at 300 K are 1.103, 1.105, 1.107 and 1.177 for HCOOH, H13COOH, DCOOH and HCOOD, respectively (Perrin et al. 2002; Baskakov et al. 1999, 2003, 2006).
2.3 Species identification, modeling and uncertainties
For the identification and modeling of individual species we adopted the same procedure as described in Jørgensen et al. (2016): since the density of lines is high enough to approach the confusion limit at different points in the spectra, it is not practical to identify all clean transitions of a specific species and, for example, create rotation diagrams based on their (measured) intensities. In fact, for a reliable identification of a given species it is important to demonstrate that there are no anti-coincidences, i.e. the inferred column density does not predict bright lines that are not observed. Also, inherent to the rotation diagram method is that the line emission is optically thin, which may not be the case for the main isotopologues of the complex organics on the scales that we consider. Rotation diagrams may therefore lead to underestimated column densities, while fainter optically thin transitions that are not considered in the fits, and for the derived column density predicted not to be observable, in fact are present at a significant level. Nevertheless, carefully constructed rotation diagrams may be useful to check the consistency of derived temperatures and column densities.
We calculate synthetic spectra for each species and compare those to the data for the most part similar to what is done in many other surveys of regions with dense spectra (e.g. Comito et al. 2005; Zernickel et al. 2012; Belloche et al. 2013, 2016; Crockett et al. 2014; Neill et al. 2014; Cernicharo et al. 2016, as well as previous PILS papers). Rather than fitting the individual lines, synthetic spectra are calculated assuming that the emission from the molecules are in local thermodynamic equilibrium (LTE), taking into account optical depth and line overlaps. With this approach, the parameters going into the modeling are the column density of the molecule, its excitation temperature, the LSR velocity, the width of the lines and the extent of the emission on the sky. These synthetic spectra can then be directly compared to the observed spectrum over the full spectral range and constrain those parameters. In this paper, the LTE assumption for the complex organic molecules is justified by the high density, 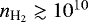 cm−3, in the environment of IRAS16293B on the 30–60 AU scales considered here, as demonstrated by the non-LTE calculations for methanol in Jørgensen et al. (2016). As noted in previous PILS papers (Coutens et al. 2016; Jørgensen et al. 2016; Lykke et al. 2017), all features toward IRAS16293B are well reproduced with a source size of 0.5″, a linewidth (FWHM) of 1.0 km s−1 and a LSR velocity of 2.5–2.7 km s−1. This likely reflects the fact that the material on the scales of the ALMA resolution is relatively homogeneously distributed and dominated by one physical component.
cm−3, in the environment of IRAS16293B on the 30–60 AU scales considered here, as demonstrated by the non-LTE calculations for methanol in Jørgensen et al. (2016). As noted in previous PILS papers (Coutens et al. 2016; Jørgensen et al. 2016; Lykke et al. 2017), all features toward IRAS16293B are well reproduced with a source size of 0.5″, a linewidth (FWHM) of 1.0 km s−1 and a LSR velocity of 2.5–2.7 km s−1. This likely reflects the fact that the material on the scales of the ALMA resolution is relatively homogeneously distributed and dominated by one physical component.
With those parameters fixed, the remaining quantities to be derived for each species are excitation temperatures and column densities for each species. The uncertainties for the typical fits can be estimated comparing synthetic diagrams to the observed spectra. Figures B.2 and B.3 in Sect. B.1 compare the best fit for CH3OD to models where the derived excitation temperatures and column densities are varied. As seen, models with either excitation temperatures or column densities higher or lower by 20% start providing fits that are visibly worse and not reproducing the observed spectra. These percentages can therefore be considered conservative uncertainties on each fit. Naturally, these two parameters cannot be considered as completely independent, but varying the excitation temperature by ±20% results in changes of the derived column densities of the species of <10%.
For rarer isotopologues with weaker lines than shown in Fig. B.3 the excitation temperature typically cannot be derived independently but that from the main isotopologue is adopted, in which case the column density is the only free parameter and the uncertainty remains the same as long as lines with signal-to-noise ratio (S/N) larger than 5 (i.e. statistical uncertainty is < 20% for the intensity of each line) are considered. Likewise, the statistical uncertainty on the line fits for species with more, and stronger, transitions may be smaller. However, those are also the species where optical depth become more important and where the range of lines start to be more sensitive to the exact structure of the emitting region – including, whether, for example, a gradient in temperature should be considered: close to the central protostars the temperatures are strongly varying and therefore the single excitation temperature derived for each species likely represents the temperature in the gas where the bulk of the emission from that species originate.
For different isotopologues of the same molecule, it is a reasonable first assumption that the excitation temperature is unchanged as long as only optically thin lines are considered. As found in previous PILS papers (e.g. Coutens et al. 2016; Lykke et al. 2017), there are some differences with respect to the optimal excitation temperatures for different groups of species. While glycolaldehyde, ethylene glycol (Jørgensen et al. 2012, 2016) and formamide (Coutens et al. 2016) are well-modeled with an excitation temperature of around 300 K, lines of other species such as acetaldehyde and ethylene oxide are best reproduced with a lower excitation temperature of around 125 K. It seems that other species follow this trend. Within the uncertainties quoted above (25 K for an excitation temperature of 125 and 60 K for a temperature of 300 K) and the small dependence of column density on those we therefore broadly group the species into “hot” (300 K) or “cold” (125 K) and use those excitation temperatures. While, in principle, small variations may be present on a species-to-species level within these groups, those variations will more likely be statistical at this level rather than representing the physical structure. Again, any differences at this level will only cause minor variations in the derived column densities. For relative abundances expressed as ratios of column densities between different species, error propagation translate the 20% uncertainties into an uncertainty of ≈ 30% – or 0.6 or 1.5 percentage points for D/H ratios of 2 or 5%, respectively. But, the error could be lower if only calibration uncertainty is an issue and other parameters such as Tex can be assumed to be the same. This should certainly be valid for the comparison between the 12C- and 13C-isotopologues and likely also some of the deuterated species.
As a comparison to our method, rotation diagrams for CH3OD and a-CH3CHDOH are shown in Fig. 1. CH3OD is an example of a species with a significant number of completely well-isolated lines spanning a range of energy levels. The errors on the derived excitation temperatures and column densities are ≈10 and 5%, respectively (taking into account the statistical noise on the derived line strengths and calibration uncertainties) with the numbers otherwise in agreement with those derived above. For a-CH3CHDOH the isolated lines represent a narrow range in energy levels, so for this species only the column density can be properly constrained. The derived uncertainty on the column density is also about 6%. This value reflects that the fitted, most isolated, transitions have S/N of 8–16, i.e. that the statistical uncertainties on their line fluxes are about 6–12% (9% on the average). When fitting just the column density by matching the intensities, the derived error goes down with the square root of the number of lines – in this case six. That is, the derived uncertainty is a factor of about 2.5 below the average statistical uncertainties of the fluxes of the individual lines. Other separate checks that these uncertainties are reasonable come from statistical fits to species with few well-isolated lines where either rotation diagrams (Jørgensen et al. 2016) or full χ 2 -analyses (Persson et al. 2016) give similar uncertainties.
Uncertainties arising from vibrational contributions to the partition functions are small (about 5% or less) for the 13C isotopologues of molecules with low excitation temperatures (~125 K) or of those for which the vibrational states are well-characterised. For deuterated species or for molecules with large correction factors, i.p., ethanol and methyl formate, this uncertainty may be up to 10–25%. Generally, the assumption that the vibrational factor of an isotopologue of a heavier molecule is the same as that of its main isotopic species will lead to an underestimate of its column density and thus lead to an estimated D/H ratio that is somewhat too small or a 12C/13C ratio somewhat too large.
 |
Fig. 1 Rotation diagrams for well-isolated lines of CH3OD (upper panel) and CH3CHDOH (lower panel). For the former, both rotation temperature and column density can be constrained, while the limited range of CH3 OD transitions only allow us to constrain the column density for that species. In both panels, the best fit is shown with the solid line with our conservative estimates on the uncertainties represented by the dotted lines (the best fit values given in the upper right corner of each panels). The dashed lines in both panels indicate the best fits assuming a fixed rotation temperature of 125 K: this temperature clearly does not match the data for CH3 OD over the range of upper energies for the observed lines. |
3 Results
Many of these detections mark the first reports of these species in the ISM, including the detections of the deuterated isotopologues of ethanol, acetaldehyde, ketene and formic acid. Other species have previously been seen toward Sgr B2(N2) (e.g. the 13C-isotopologues of ethanol by Müller et al. 2016) or Orion KL (e.g. CH2DOCHO by Coudert et al. 2013) and a few have been identified from single-dish observations of IRAS16293 (i.e. methyl formate, CH3OCDO, tentatively by Demyk et al. 2010, and dimethyl ether, CH2DOCH3, by Richard et al. 2013). However, for those species the systematic study presented here allows for the first time a direct comparison between these species with a high number of identified lines originating in a region that is spatially resolved on solar system scales and with what is thought to be under relatively homogeneous physical conditions in terms of temperatures and densities.
3.1 Emission morphology
The bulk of the analysis in this paper concerns the identification of lines toward single positions, but one of the main strengths of the ALMA data is to provide fully sampled images. Therefore a few words about the emission morphology are included here.
As discussed in Jørgensen et al. (2012, 2016), the emission around IRAS16293B is complex: toward the location of the protostar itself the emission from the dust continuum as well as that of many lines is optically thick. This causes many of the lines in the spectra to appear in absorption hampering interpretation. For identification and modeling of species, it is instead more useful to consider positions offset by 0.5–1.0″ from the continuum peak where the line emission from many of the complex organic molecules appears brighter and where absorption is less of an issue.
Figure 2 compares the integrated emission around IRAS16293B for two transitions of CH OH and CH2DOH representing different excitation levels. This highlights that the emission morphologies are remarkably similar for all species and lines of different excitation levels, what is also seen for other complex organic species identified from the PILS data (Jørgensen et al. 2016; Coutens et al. 2016; Lykke et al. 2017). This reenforces the point above that the bulk of the emission is dominated by relatively compact material with relatively homogeneous physical conditions.
OH and CH2DOH representing different excitation levels. This highlights that the emission morphologies are remarkably similar for all species and lines of different excitation levels, what is also seen for other complex organic species identified from the PILS data (Jørgensen et al. 2016; Coutens et al. 2016; Lykke et al. 2017). This reenforces the point above that the bulk of the emission is dominated by relatively compact material with relatively homogeneous physical conditions.
Also marked in Fig. 2 are the continuum peak and the two offset positions at one half and one full beam (0.5′′ and 1′′): the emissionpeaks close to the half-beam offset position with its strength reduced by a factor of 2 at the full beam offset position. This is consistent with the emission peak being marginally resolved in that direction. Fig. 3 compares spectra toward the two offset positions over a frequency range where prominent (low excitation) lines of CH2DOH are seen. The figure also shows a synthetic spectrum calculated at low temperatures indicating the location of the CH2DOH transitions. It is clearly seen that many of these lines that show absorption at the half-beam offset position are in emission at the full beam offset position. Indeed, the synthetic spectrum predicts that these specific transitions will be optically thick (τ > 1).
In fitting the data, we therefore iteratively selected transitions that have τ < 0.1. Focusing only on the optically thin transitions, it is found that there is about a factor of 2 difference between the derived column densities (assuming the same filling factor) toward the full- and half-beam offset positions. Given that the emission is only marginally resolved, this likely reflects a combination of the actual drop in column density of the species as well as the fact that the emission does fill a smaller fraction of the beam at the full-beam offset position (Fig. 2). In the following work, we therefore predominantly focus on the full beam position, but note (i) that we check for consistency of the fits toward the half-beam position and (ii) that for comparison to other results from the half-beam positions, one can multiply the quoted column densities by a factor of 2.
 |
Fig. 2 Representative maps for lines of CH |
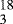
 |
Fig. 3 Spectrum of optically thick transitions of CH2DOH at 356.95 GHz toward the half-beam (upper) and full-beam (lower) offset positions from IRAS16293B. In the lower panel, synthetic spectra for CH2DOH are overlaid:those are both calculated for the same column density (Table 1) but with temperatures of 50 K (blue) and 300 K (red). |
3.2 Derived column densities
Figures B.1–B.26 show the observed spectra for each of the species, compared to the predictions by the synthetic spectra and the individual fits are discussed in the appendix. The data show detections of each of the (singly) deuterated versions as well as the 13C isotopologues of the targeted oxygen-bearing complex organic molecules (the 13C isotopologues for ethanol and methyl formate only tentatively).
Table 1 lists the derived column densities for each of the species and, for the rarer isotopologues, the abundances relative to the main isotopologue. The column densities of the deuterated species range from 2% up to 18% relative to the main isotopologues. A significant part of this variation is due to the composition of the functional groups. For the same D/H ratio, one should expect a molecule where the deuterium is substituted into one of the hydrogen atoms in the methyl CH3 group to be three times more abundant than a molecule where the substitution is into a functional group such as OH with only one hydrogen atom. If this effect is taken into account, surprisingly small variations are seen in the D/H ratios for the individual isotopologues. A distinction is apparent with the simpler molecules consistent with a D/H ratio of ≈2%, and the more complex species methyl formate, acetaldehyde, ethanol and dimethyl ether show higher ratios ranging from about 4 to 8%, similar to the values of glycolaldehyde. For dimethyl ether a lower 12C/13C ratio is found, about half that of the standard ratio for the local ISM (68; Milam et al. 2005), similar to what has previously been inferred for glycolaldehyde. The marginal cases of 13C isotopologues of methyl formate and ethanol are also consistent with lower 12C/13C ratios but additional observations are needed to confirm and solidify the numbers for these two species.
Derived column densities for the isotopologues of the complex organics and abundances relative to the main isotopologue at the position offset by 0.5″ (60 AU) from IRAS16293B.
4 Discussion
The presented analysis provides important clues concerning the origin of various complex organic molecules in different ways. As summarised in Fig. 4, the species fall in a number of different groups depending on their excitation temperatures and the deuteration levels. In this section we try to reconcile these observations to discuss the implications for formation of the different complex organics.
 |
Fig. 4 Schematic representation of the results from this paper: the discussed molecules are grouped according to their D/H ratios and excitation temperatures. The species underlined can be shown to have lower 12C/13C ratios by a factor 2 than typical values for the local ISM (although only tentatively for methyl formate and ethanol), while the remainder are either consistent with a standard ISM ratio or have too optically thick lines of the main isotopologuefor a reliable independent estimate. |
4.1 Excitation temperatures
An important aspect of the presented analysis is the variations in the excitation temperatures between the different species with one group consistent with temperatures of approximately 100–150 K and one with temperatures of 250–300 K. The former group includes CH2CO, CH3CHO, CH3OCH3 as well as H2CO (Persson et al. 2018) and c-C2H4O (Lykke et al. 2017), while the latter group includes CH3OH, C2 H5OH, CH3OCHO as well as NH2CHO and HNCO (Coutens et al. 2016)and CH2OHCHO and (CH2OH)2 (Jørgensen et al. 2016). These divisions are mostly in agreement with a similar division into “cold” (Tex ≲ 100 K) and “hot” (Tex ≳ 100 K) molecules by Bisschop et al. (2007) from a survey of high-mass hot cores and a follow-up study by Isokoski et al. (2013). Also, the molecules with low excitation temperatures are typically those identified in the large single-dish survey of complex organics toward IRAS16293–2422 by Jaber et al. (2014). Table 2 compares these detections: in general it seems that the hot and cold molecules from the high-mass surveys and the interferometric data agree well with a few exceptions. Also, all the cold molecules from the interferometric data are detected in the IRAS16293 single-dish survey again illustrating that that survey may be biased toward the colder gas on larger scales. Of the warm molecules in the interferometric data, CH3OCHO, CH3OH and NH2CHO are seen in the single-dish survey: those are all relatively abundant and thus may contribute significantly enough to be picked out in the larger beam. None of the organics that were detected in the single-dish observations by Jaber et al. (2014) remain undetected in the interferometric data presented in this paper.
As pointed out previously (e.g. Garrod et al. 2008), a straightforward explanation for the distinction between hot and cold molecules may lie in the binding energies of the different species (also listed in Table 2). A very clear distinction is seen between species that have high excitation temperatures in IRAS16293 interferometric data, which have binding energies of 5000–7000 K (ethylene glycol as high as 10 200 K), and those that have low excitation temperatures, which have binding energies in the 2000–4000 K range. In the context of the three-phase chemical model by Garrod (2013), this would imply that the bulk of the colder species simply sublimate at lower temperatures and vice versa for the warm species: the lower binding energies would correspond to sublimation temperatures of ≈50 K and the higher ones to ≈100 K. One should note though that these binding energies are for pure ices. If the species are mixed in ice with H2 O or CH3OH the binding energies would increase to the values of those, ≈5500 K.
The difference between these (lower) sublimation temperatures of 50–100 K and the higher inferred excitation temperatures (100–300 K) may be a result of the rapid infall (and short distances) in the dense innermost region of the envelope around IRAS16293B. For a typical infall speed of ~1 km s−1, the time it would take material to move ~50 AU (typical of these temperature regimes in the IRAS16293) is ~200 years. Thus, while the separation of the molecules into “hot” and “cold” species likely reflects where those species originally sublimate, this short timescale would imply that the derived values for the excitation temperatures reflect the current location of the bulk of the species closer to the protostar. As argued by Schöier et al. (2002), this short timescale may also inhibit any “second generation” formation of complex organics, at least based on those species sublimating only at the higher temperatures. The ambiguity for dimethyl ether may reflect that this species toward the high-mass sources can be formed by “second generation” chemistry through gas-phase reactions (e.g. Charnley et al. 1992) as is suggested by the multiple components observed toward the multi-line study of this species toward the high-mass hot core G327.3−0.6 (Bisschop et al. 2013).
Division of molecules into “hot” and “cold” from the interferometric observations of IRAS16293 (this study) and high-mass hot cores (Bisschop et al. 2007), an indication of whether or not the species are detected in the single-dish study of IRAS16293 (Jaber et al. 2014) and the binding energies of each species (from the tabulation by Garrod 2013).
4.2 Deuteration
4.2.1 Observed ratios
The analysis presents new accurate measurements for the D/H ratios of complex organics representative for the warm inner envelope T >100 K of IRAS16293B where most of the molecules have sublimated off the dust grains. For the species considered in this paper, and for glycolaldehyde, isocyanic acid and formamide (Jørgensen et al. 2016; Coutens et al. 2016), no differences are seen between the fractionation in different functional groups. It is therefore possible to assign one value per species (after taking the statistics for species with multiple H atoms into account). This is summarised in Table 3 and compared to the single-dish estimates from the literature.
The three most important takeaways from this comparison are (i) the consistency between D/H ratios on the ~ 50 AU scales probed by the ALMA observationsand those on ~1000 AU scales probed by the single-dish data (except for H2O), (ii) the small, but significant variations between the interferometric measurements for the different species and (iii) the lack of variations in the deuteration for the different functional groups.
The strength of this study, besides providing new detections for a number of deuterated isotopologues, is the systematic census. Previous (single-dish)studies have typically focused on brighter lines from one or two species and, as noted above, care must be taken with estimates of column densities for the main isotopologues, in particular, due to optical depth issues. Also, the larger number of lines for many of the observed isotopologues means that the accuracy of the absolute column density determinations are at the 10–20% level and relative abundances between isotopologues of individual species likely better than that. Differences of factors of 1.5–2.0 in the D/H ratios in Table 3 are therefore significant. Table 3 still shows that there is a good agreement between the single-dish and interferometric D/H ratios with the exception of water, for which the interferometric value is a factor of 5 lower than that based on single-dish measurements.
It appears that there are some systematic variations between the organics: methanol, ketene, formic acid all have D/H ratios of approximately 2% similar to formamide and isocyanic acid (Coutens et al. 2016) and formaldehyde (Persson et al. 2018). Dimethyl ether has a D/H ratio of ≈4%, ethanol, methyl formate and glycolaldehyde ≈5–6% and acetaldehyde the highest ratio of ≈8%. Water shows much lower D/H ratios than the organics, likely reflecting its formation in the earlier prestellar stages where the degree of deuteration is lower (e.g. Taquet et al. 2014; Furuya et al. 2016).
Compared to the study of the Sgr B2(N2) by Belloche et al. (2016), the detections of the deuterated isotopologues of ethanol and methyl formate are new and all ratios for the low-mass IRAS16293 are higher than those for the high-mass source. For methanol, our CH2DOH measurement is a factor of 50 above theestimate for CH2DOH of Belloche et al. (2016) and our CH3OD estimate more than a factor of 100 above the upper limit for Sgr B2(N2). The derived column density ratios for the different deuterated ethanol variants relative to the main isotopologue is a factor of 2–4 above the upper limitstoward Sgr B2(N2). The column density ratio for one variant of deuterated methyl formate, CH2DOCHO, is about a factor of 7 higher than the upper limit of Belloche et al. (2016).
It is interesting to note that there is a lack of variation in the D/H ratios for the different functional groups, so that, for example, the [CH2DOH]/[CH3OD] ratio is very close to a factor of 3 as expected from statistics. This is different from what is found in prestellar cores with much higher ratios ≳10 (e.g. Bizzocchi et al. 2014) or high-mass star-forming regions such as Orion KL with lower ratios ~ 1 (e.g. Jacq et al. 1993, Peng et al. 2012, Neill et al. 2013). Such differences from the statistical ratios led Faure et al. (2015) to propose that exchange reactions between the -OH groups of H2O and CH3OH during the warm-up phase could significantly alter the relative abundance ratios. This in turn would predict an inverse scaling between the [CH2DOH]/[CH3OD] ratio on the one hand and the [HDO]/[H2O] ratio on the other. Also, they noted that the previous high [CH2DOH]/[CH3OD] ratios reported based on single-dish observations for IRAS16293 compared to those in Orion KL could reflect the presence of heterogeneous ices with respect to D/H ratios in IRAS16293. However, as noted above revised methanol values from the single-dish data are in agreement with that inferred from our analysis here. Also, the revised measurements of the [HDO]/[H2O] ratio toward IRAS16293 from interferometric measurements (Persson et al. 2013) representing the fully sublimated ice mantles, is lower than what was adopted in Faure et al. (2015). Faure et al. (2015) quote a timescale of ~ 103 years for equilibrium in their calculations, which may be too long given the short dynamical timescale governing the infall of material in the inner envelope of IRAS16293 as noted above. It is possible that exchange reactions could play a role in the upper layers of the ice mantles reflecting the higher D/H ratios of water measured in the ambient cloud (Coutens et al. 2012) or that CH3OH and H2O simply are not co-located (Cuppen et al. 2009).
Inferred abundances and deuterium fractionation (from interferometric and single-dish data, respectively) for different organic molecules.
4.2.2 Comparison with models
A qualitative comparison can be made to the models by Garrod (2013). Those models followed the formation of complex organic molecules through radical–radical recombination reactions taking place during the warm-up of the core surrounding of a high-mass protostar. When looking at the species in those models, it isinteresting to note that the species with D/H ratios of 2% (formaldehyde, methanol and ketene) are all abundant in the ices from the very beginning of the warm-up, ethanol and dimethyl ether production in those models set in at temperatures around 20 K, while methyl formate and glycolaldehyde only appear in the ices at temperatures ≳ 25 K. The same models also predict significant isocyanic acid ice present at low temperatures, again consistent with its low D/H ratio of ≈1% (Coutens et al. 2016) and some formamide ice is formed through reactions between NH2 and H2 CO already at low temperatures as also seen in recent laboratory experiments (Fedoseev et al. 2015, 2016; see also the discussion in Coutens et al. 2016). Formic acid, HCOOH, is the only species that seems to be an exception of this trend. In the models of Garrod (2013), formic acid predominantly forms in the gas phase when formaldehyde evaporates off dust grains and then freezes out on the dust grains immediately. However, Taquet et al. (2014) point to recent laboratory and theoretical studies (Goumans et al. 2008; Ioppolo et al. 2011) and argue that formic acid more likely is formed in the cold ices through reactions between CO and OH. In either case, it is not unreasonable to expect the ≈2% D/H ratio for formic acid observed, either because it is inherited from the formaldehyde or because it is set in the early phases in the ices together with most of the other species.
A directquantitative comparison can be made to the results of Taquet et al. (2014) who presented an extensive model for the formation and deuteration of interstellar ices during the collapse of a protostellar core including also the sublimation of these species closeto the protostar. In those models, species are predominantly formed on ice surfaces during the warm-up or collapse of material in the pre- and protostellar core and thereby typically inherit the D/H ratios at that specific time or layer in the ices. This means that the species forming later or in the outer parts of the ices would be characterised by higher D/H ratios than those formed earlier/deeper in the ices. Also, in those models, the D/H ratios for individual functional groups should be correlated across the different species, reflecting the formation pathways based on the radical recombinations.
The models of Taquet et al. (2014) predict lower D/H values on smaller (interferometric) than larger (single-dish) scales, but as noted above this is not supported by the current observations, except for water. Still, care must be taken that although the single-dish observations are weighted toward the gas on larger scales, the strong enhancement of the species closeto the two components of IRAS16293 will weigh heavily there. Figure 5 compares the measured D/H ratios to the predictions on interferometric scales at three separate times after collapse – 1.1 × 105 years (“early”), 1.5 × 105 years (“intermediate”) and 2.0 × 105 years (“late”) – in the models by Taquet et al. (2014). The D/H ratios (corrected for statistics) are sorted by species (left) and radical/functional group determining the D/H ratio (right). Generally it is seen that while the “late” model significantly under-predicts the observed D/H ratios for each species (predictions of order 0.1% vs. the observed few %), the “early” and “intermediate” models provide a much better agreement, although, in particular, the “early” model slightly overestimates the observed values (with a few exceptions, i.p., methanol). The “early” model predicts large variations between the D/H ratios for different functional groups for individual molecules (in particular, ethanol and glycolaldehyde), which is not observed. These variations become smaller in the “intermediate” and “late” models. Taken together these comparisons, in particular, the slightly overprediction of the D/H ratios in the “early” model compared to the observations and more homogeneous functional group D/H ratios at late times, could be taken as an indication that the biggest challenge in fact is to determine the initial conditions and/or time in the models that applies best in the specific case.
A slightly different paradigm to those presented by Garrod (2013) and Taquet et al. (2014) has been offered by Drozdovskaya et al. (2015) who modeled the 2D structure of the complex organic chemistry of a static protostellar envelope with a significant outflow cavity. The introduction of such an outflow cavity would significantly alter the chemistry by allowing dust to be heated and FUV photons escaping to larger scales leading to enhanced photodissociation. Drozdovskaya et al. (2015) demonstrated that this would lead to a time ordering of the appearance of complex organic molecules (their Fig. 11) with, in particular, ethanol and dimethyl ether forming during the protostellar stages where the FUV photons from the young protostar enhance the radicals resulting in their formation, compared to the other species being formed during the prestellar core stage. Such a time-ordering would thus imply a three-step formation of the complex organics: formaldehyde, methanol, ketene and formic acid would be formed early in the prestellar core where the D/H ratio is low, acetaldehyde, methyl formate and glycolaldehyde at the late prestellar/early protostellar stage where the D/H ratio reaches its maximum and dimethyl ether and ethanol in the protostellar envelope when the FUV field becomes important.
In either scenario the distinct formation time of groups of species may also reflect in the resulting 12C/13C ratios: as discussed in Jørgensen et al. (2016) and noted in Sect. 1, a lower 12C/13C ratio than the ISM value may reflect fractionation or differences in sublimation temperatures of, i.p., 12CO and 13CO. A possible difference in the sublimation temperatures for 12CO and 13CO may play a role at the later stages when the protostellar heating is starting to increase the temperature to the point when CO starts sublimating. Smith et al. (2015) demonstrated that small differences in the binding energies between 12CO and 13CO (the latter more tightly bound) of up to 10 K could lead to an enhanced 12CO:13CO gas-phase abundance ratio by up to a factor of 2 observed at infrared wavelengths toward some sources. This would naturally have the opposite effect of the 12C/13C of the organics derived from the CO “left behind in the ices” that would be deficient in 12C relative to 13C. These effects would mainly play a role in the region where the temperature is just around what is required for CO to sublimate and thus mainly affecting species formed at the onset of warming-up by the protostar itself. Fractionation due to the ambient UV field in contrast is unlikely to introduce any significant differences as it predominantly occurs during the early cloud stages: an anomalous 12C/13C ratio for specific species therefore would have to be carried through to their formation time of without affecting the other species. However, if in fact the formation of some of the complex organics require the FUV irradiation provided by the central protostar such as in the scenario described by Drozdovskaya et al. (2015), this could perhaps trigger an anomalous 12C/13C ratio for some species.
Clearly, more comparisons like these are needed to move forward and test the different scenarios. This could, for example, be through similar comprehensive comparisons for more sources but also to other predictions in the models (e.g. the absolute abundances for the individual organics). Such comparisons would, in particular, make it possible to address the question of whether the source physical evolution during the early stages is the critical parameter in determining the D/H ratio. Also, observationsat higher angular resolution should be able to resolve the emission regions of the groups of species with different excitation temperatures and thus test that spatial segregation. In this context, it would naturally be interesting to see whether the relatively high excitation temperatures inferred for IRAS16293B and some high-mass hot cores relative to the Galactic center apply in general, whether they reflect the distribution of the material around those sources or whether, for example, additional production of some species at high temperatures come into play.
 |
Fig. 5 Comparison between the D/H ratios (per H-atom) for the complex organics sorted by species (left panel) and the leading radical in the formation (right panel). For each species, the PILS observations are represented by the first, red, bar and the predictions in the models by Taquet et al. (2014) after 1.1 × 105, 1.5 × 105 and 2.0 × 105 years by the second (blue), third (orange) and fourth (green) bars, respectively (see also legend in the part of the right panel). For the model at 2.0 × 105 years, the D/H ratios have been multiplied by a factor of 10 to enable comparisons. |
5 Conclusions
We have presented a systematic survey of the isotopic content of oxygen-bearing (and some nitrogen-bearing from Coutens et al. 2016)complex organic molecules toward the low-mass protostar IRAS16293B. Using data from the ALMA Protostellar Interferometric Line Survey (PILS) program, we provide an inventory of those species and their relative abundances in the warm gas close to the central protostar where ices are completely sublimated. The main conclusions are as follows:
For the first time we identify the deuterated and 13C-isotopic species of ketene, acetaldehyde and formic acid as well as deuterated ethanol in the interstellar medium and present observations of 13C isotopic species of dimethyl ether, methyl formate and ethanol (the latter two only tentatively detected) along with mono-deuterated methanol, dimethyl ether and methyl formate. Systematic derivations of excitation temperatures and column densities result in small uncertainties in their relative abundances.
Some differences are found in excitation temperatures for different species, with the lines for one group (formaldehyde, dimethyl ether, acetaldehyde and formic acid, together with formaldehyde; Persson et al. 2018), best fit with an excitation temperature of approximately 125 K with the remaining species, together with formamide and glycolaldehyde (Coutens et al. 2016; Jørgensen et al. 2016), better fit with a temperature of 300 K. This possibly reflects different binding energies of the individual species to the grains with the former group having binding energies in the 2000–4000 K range and the latter in the 5000–7000 K range. The division is very similar to what has previously been found for high-mass sources (Bisschop et al. 2007).
The D/H ratios of the complex organic molecules can be divided into two groups with some of the simpler species (methanol, ketene, formic acid) as well as formaldehyde, formamide and isocyanic acid showing D/H ratio of ≈2% and the more complex species dimethyl ether, ethanol, methyl formate, glycolaldehyde and acetaldehyde showing higher ratios ranging from about 4 to 8%. Conservative estimates of the errors on these ratios are 0.6 percentage points for the lower value and 1.5 percentage points for the higher values. This distinction may reflect the formation time of each species in the ices before or during warm-up/infall of material through the protostellar envelope.
No significant differences are seen in the deuteration of different functional groups for individual species, possibly a result of the short timescale for infall through the innermost regions where exchange reactions between different species may be taking place.
-
The 12C/13C ratio of dimethyl ether is found to be lower than that of the local ISM, similar to the case of glycolaldehyde (Jørgensen et al. 2016). Marginal detections of the 13C isotopologues of methyl formate and ethanol are also consistent with a lower ratio. Low 12C/13C ratios may reflect the formation histories of individual species; that they form in the ices at a late point where more 13C is available due to a slightly lower binding energy of 12CO compared to 13CO ice – or fractionation triggered by FUV irradiation from the central protostar.
This study is an important illustration of how the isotopic composition can be used to determine the formation pathways for different molecular species. Efforts on analysing the nitrogen-containing organics toward IRAS16293B (Ligterink et al. 2017, 2018; Coutens et al. 2018; Calcutt et al. 2018), as well as establishing comparable inventories of both oxygen- and nitrogen-bearing species toward IRAS16293A, are ongoing. Together these studies will serve as an important constraint on astrochemical models attempting to account for the fractionation processes in protostellar environment. It would be worthwhile revisiting the abundances of species on larger scales for IRAS16293 and other sources, given the issues with opacity effects for many of the main isotopologues. As noted above, additional efforts are needed to investigate any deviations of the 12C:13C ratios for the organics that are only marginally detected or those suffering from optical depth issues. Finally, extending this survey to other embedded protostars, possibly in other regions, could shed light on the importance of environment versus evolutionary histories of the sources for the formation of complex molecules.
Acknowledgements
The authors wish to thank the referee for good, constructive comments that significantly improved the paper. The authors are grateful to the various spectroscopy groups without whose systematic efforts, studies like this would not be possible. In particular, thanks go to Adam Walters and Frank Lewen for their contributions in Cologne and Laurent Margulès and Roman Motiyenko for their work in Lille. This paper makes use of the following ALMA data: ADS/JAO.ALMA#2013.1.00278.S. ALMA is a partnership of ESO (representing its member states), NSF (USA) and NINS (Japan), together with NRC (Canada), NSC and ASIAA (Taiwan), and KASI (Republic of Korea), in cooperation with the Republic of Chile. The Joint ALMA Observatory is operated by ESO, AUI/NRAO and NAOJ. The group of J.K.J. is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme through ERC Consolidator Grant “S4F” (grant agreement No. 646908). Research at Centre for Star and Planet Formation is funded by the Danish National Research Foundation. A.C. postdoctoral grant is funded by the ERC Starting Grant 3DICE (grant agreement 336474). M.N.D. acknowledges the financialsupport of the Center for Space and Habitability (CSH) Fellowship and the IAU Gruber Foundation Fellowship. The group of EvD acknowledges ERC Advanced Grant “CHEMPLAN” (grant agreement No. 291141).
Appendix A Discussion of individual species
A.1 History of detections
Methanol, CH3OH, (Ball et al. 1970), acetaldehyde, CH3CHO, also known as ethanal (Gottlieb 1973; Fourikis et al. 1974), dimethyl ether, CH3OCH3 (Snyder et al. 1974), ethanol, C2 H5OH (Zuckerman et al. 1975), methyl formate, CH3OCHO (Brown et al. 1975; Churchwell & Winnewisser 1975) and also ketene, CH2CO, also known as ethenone (Turner 1977) were all among the early molecules detected by means of radio astronomy in the 1970s. All detections were made toward high-mass star-forming regions, mostly in the prolific Galactic Centre source Sagittarius B2. Methanol and ethanal were detected also in Sagittarius A; dimethyl ether was detected in the Orion Molecular Cloud. Unambiguous detections of minor isotopic species have also been reported for several of these molecules: The 13C (Gottlieb et al. 1979)and 18O (Gardner et al. 1989) species of methanol were detected, and in the case of deuterated isotopologues even CD3OH (Parise et al. 2006). In the case of methyl formate both 13C isotopologues were detected, along with CH3OCDO (tentatively; Demyk et al. 2010), CH2DOCHO (Coudert et al. 2013) and both 18O isotopologues (Tercero et al. 2012). Detections of deuterated and 13C dimethyl ether were reported by Richard et al. (2013) and Koerber et al. (2013), respectively, while the detection of the 13C isotopomers of ethanol toward Sagittarius B2 were reported by Müller et al. (2016). Formic acid was detected first toward Sgr B2 (Zuckerman et al. 1971; Winnewisser & Churchwell 1975). It was also detected in a cold cloud (Irvine et al. 1990) and toward IRAS16293 (Cazaux et al. 2003). To our knowledge, there are no secure detections of minor isotopic species of HCOOH.
A.2 Methanol, CH3OH
As mentioned above, one of the biggest problems about fitting CH2DOH is that many of its transitions become optically thick at low temperatures. For the first we therefore only utilise lines that are predicted to have τ < 0.1 and further indicate additional lines that are strongly optically thick at either 50 or 300 K separately. Those lines are typically overproduced in the synthetic spectra here, likely because their emission is quenched by the colder material at larger scales. Three transitions (one seen at 349.62 GHz and two at 346.82 GHz) are still significantly overproduced by the models. This likely reflects issues with the catalogue entries for these high excitation transitions. The fits to CH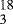 OH at the half beam offset position were discussed in Jørgensen et al. (2016). For completeness, Fig. B.5 shows the fits at the one beam offset position where the derived column density is taken to be a factor 2 lower than at the half beam offset position. The CH3OD lines are well-reproduced for both the a- and e-type transitions, although these lines span a more limited set of upper energy levels than for many of the other species. For all methanol transitions, an excitation temperature of 300 K works well. For CH
OH at the half beam offset position were discussed in Jørgensen et al. (2016). For completeness, Fig. B.5 shows the fits at the one beam offset position where the derived column density is taken to be a factor 2 lower than at the half beam offset position. The CH3OD lines are well-reproduced for both the a- and e-type transitions, although these lines span a more limited set of upper energy levels than for many of the other species. For all methanol transitions, an excitation temperature of 300 K works well. For CH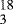 OH and CH3OD, 36 and 29 lines can be assigned, respectively.
OH and CH3OD, 36 and 29 lines can be assigned, respectively.
A.3 Ethanol, CH3CH2OH
For the main isotopologue of ethanol similar issues as for CH2DOH arise with lines that become optically thick at larger scales. Those are indicated in the figures. Still, more than hundred lines of ethanol are predicted to be optically thin above 5σ. The lines ofthe different deuterated variants vary somewhat in strength: for the two variants of CH2DCH2OH and CH3CHDOH >60 and 37 transitions are identified, respectively providing reliable estimates of their column densities. For CH3CH2OD about 5–10 features can be assigned: these are blended to varying degree making the assignment more ambiguous and the derived column density tentative. As for methanol, the lines of ethanol are well-produced with an excitation temperatureof 300 K but it could be 10–20% higher or lower without significantly altering the fits. For the two 13C isotopologues a few plausible assignments are seen in the spectra. Individually they are not sufficient to claim a secure detection of any of the 13C isotopologues, however, if they are considered together about a handful of lines can be modeled with a column density corresponding to a 12C/13C ratio lower by about a factor 2 than the canonical ISM value.
A.4 Methyl formate, CH3OCHO
The lines of methyl formate are well-fit with an excitation temperature of 300 K. For the main isotopologue and CH2DOCHO several hundreds of lines are present above 5σ (about half of those of the main isotopologue are optically thin), while only about 10–15 lines are seen of the rarer CH3OCDO isotopologue. The implied D/H ratio derived from this variant is close to that of the CH2DOCHO isotopologue corrected for statistics, however, lending further credibility to the assignment. The lines of the 13C isotopologue are relatively faint with most of the lines at the 5–10σ levels blended to varying degrees. The best fit column density imply a 12C/13C abundance ratio of ≈30 but with large uncertainties.
A.5 Ketene, CH2CO
The analysis of ketene is based on the 13C and singly deuterated isotopologue and we present tentative detections/upper limits for the 18O and doubly deuterated isotopologue. For the main isotopologue of ketene only one out of the fourteen transitions predicted in the observed range is optically thin. The column density is therefore derived based on the 13C isotopologues and assuming a standard 12C/13C ratio. These species are best fit with an excitation temperature of 125 K. Should it turn out that the 12C/13C ratio is lower for ketene, similar to the cases of glycolaldehyde and methyl formate, the D/H ratio inferred here would be an underestimate by that factor. We also show the fits for brightest predicted lines for the 18O and doubly deuterated isotopologues: for the 18O isotopologue we assume a standard 16 O:18O ratio of 560 (Wilson & Rood 1994) from the abundance of the main isotopologue inferred from the 13C species above. The data are consistent with this fit, implying that the column density of the main isotopologue is not stronglyunderestimated, but the transitions are not sufficiently strong to consider a (tentative) detection. The doubly deuterated variant is not seen with an upper limit relative to the main isotopologue of 0.2% (< 10% relative to the singly deuterated version corrected for statistics).
A.6 Dimethyl ether, CH3OCH3
Like for ketene, but in contrast to its isomer ethanol, dimethyl ether is best fit with low excitation temperature of 125 K. The deuterated version is naturally highly abundant due to the larger number of indistinguishable H-atoms (six). For both the main and deuterated isotopologues about 100 transitions can be assigned. About 30 lines are present at the 5σ level for the 13C isotopologue: those lines are also well-fit with this excitation temperature. The ratio of the column densities between the 12C and 13C isotopologues is a factor 17, which, corrected for the statistics (13CH3OCH3 and CH3O13CH3 again being indistinguishable), implies a 12C/13C ratio of 34, about half that of the canonical ISM value and close to those measured for glycolaldehyde, methyl formate and ethanol.
A.7 Acetaldehyde, CH3CHO
Acetaldehyde is the third species best fit with a low excitation temperature (see also Lykke et al. 2017). Like for the other species a number of transitions of the main isotopologue are optically thick, but still more than 100 optically thin transitions are present at the 5σ level allowing a good estimate of its column density. For the 13C isotopologues a sufficient number of lines (about 60 and 80 transitions for CH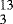 CHO and 13CH3CHO, respectively) are present to allow for a direct measurement of the 12C/13C isotope ratio, which is found to be consistent with that of the local ISM. Spectroscopic data only exists for the deuterated isotopologue with the D substituted on the -CHO group. This isotopologue is clearly detected.
CHO and 13CH3CHO, respectively) are present to allow for a direct measurement of the 12C/13C isotope ratio, which is found to be consistent with that of the local ISM. Spectroscopic data only exists for the deuterated isotopologue with the D substituted on the -CHO group. This isotopologue is clearly detected.
A.8 Formic acid, HCOOH
For a reliable estimate of the column density of formic acid we rely on lines of both the main and 13C isotopologues: the stronger lines of the main isotopologue of (trans-)formic acid are strongly optically thick (about half of the 30 lines above 5σ). We therefore utilise the 17 lines of the 13C isotopologueto derive a first estimate of the column density and from that simulate the spectrum of the main isotopologue scaled by the standard ISM ratio. Utilising this column density and focusing only on the fainter lines of the main isotopologue, a number of assignments can be thereby be made confirming the derived column density. In Fig. B.23 one line is seen at 338.202 GHz, which is clearly over-predicted by the model. That line is an example of one of the transitions being strongly optically thick (τ ≳ 1). The line at 338.192 GHz in that specific panel is centered on is optically thin (τ ~ 0.01), though. Lines of the two deuterated isotopologues are detected as well, but a number show severe blending. We consequently derive a common column density for both species based on the about 30 lines present above 5σ. It seems that such a column density does reproduce the lines of both species equally well, but the problems with the fits does make this numbermore uncertain. The deuterated isotopologues are fit assuming the same column density for both variants.
The lines of formic acid are in general well-produced with a high excitation temperature of 300 K. In addition to the trans-formic acid, we also checked for the presence of the high energy cis conformer that has recently been seen with relatively high abundances relative to the trans conformer toward the Orion Bar (Cuadrado et al. 2016), likely a result of the strong UV field there leading to photo-switching between the two conformers. In our data the cis conformer remains undetected down to an abundance of ~ 0.1% with respectto the trans conformer, which would be consistent with the expected ratio at equilibrium at the temperature of IRAS16293B. It does mark a difference though from the recent tentative detection of one line of the cis conformer toward the dark cloud B5 (Taquet et al. 2017).
Appendix B Spectra
On the following pages the observed and modeled spectra are shown for the lines predicted to be the brightest and optically thin according tothe synthetic spectra for each invidual species.
B.1 Methanol
 |
Fig. B.1 The15+16 brightest lines of CH3OD (a-type in the upper panels and e-type in the lower panels) toward the full-beam offset position. The lines are sorted according to Eup given in K in the upper left corner of each panel. The windows are centered on these specific lines. |
 |
Fig. B.2 As in Fig. B.1, zoomed-in to highlight the fits. Overplotted are the best fit models using excitation temperatures 20% above (dotted) and 20% (dashed) below the best fit value. |
 |
Fig. B.3 As in Fig. B.1, zoomed-in to highlight the fits. Overplotted are the best fit models using column densities 20% above (dotted) and 20% (dashed) below the best fit value. |
 |
Fig. B.4 The24 brightest optically thin lines of CH2DOH as expected from the synthetic spectrum toward the full-beam offset position. In the cases, where other lines fall in the band that are strongly optically thick at 50 or 300 K, they have been marked with a plus-sign or cross, respectively. The discrepancies for the lines at 349.62 and 346.82 GHz likely reflect issues with the catalogue entries for these high excitation transitions. |
 |
Fig. B.5 As in Fig. B.1, 21 brightest lines of CH |
B.2 Ethanol
 |
Fig. B.6 As in Fig. B.1, 32 brightest lines of ethanol as expected from the synthetic spectrum toward the full-beam offset position. |
 |
Fig. B.7 As in Fig. B.1, 16 brightest lines of a-13CH3CH2OH (upper panels) and a-CH |
 |
Fig. B.8 As in Fig. B.1, 16 brightest lines of a-CH3CHDOH toward the half-beam (upper panel) and full-beam (lower panel) offset positions. The asterisks next to the upper energy level in some panels indicate features for which the multiple transitions (from the same species) overlap. |
 |
Fig. B.9 As in Fig. B.1, a-CH3CH2OD the upper panels show the spectra toward the half-beam position and the lower panels the full-beam (lower) offset position. |


B.3 Methyl formate
 |
Fig. B.13 As in Fig. B.1, 16 + 16 brightest lines of deuterated methyl formate: DCOOCH3 in the upper panels and CH2DOCHO in the lower panels. |
 |
Fig. B.14 As in Fig. B.1, 16 brightest lines of 13C methyl formate, CH3O13CHO toward the half-beam (upper panel) and full-beam (lower panel) offset positions. |
B.4 Ketene
 |
Fig. B.15 As in Fig. B.1, the brightest lines of the 13C-isotopologues of ketene with 13CH2CO shown in the upper panels and CH213CO in the lower panels. |
 |
Fig. B.16 As in Fig. B.1, 16 brightest lines of the singly deuterated-isotopologues of ketene CHDCO (upper panels). The lower panels show the eight brightest lines of doubly deuterated ketene, CD2 CO (left panel) and the 18O isotopologueCH2C18O (right panel). For those we assume the same D/H ratio as for the singly deuterated species represents an upper limit/tentative identification. |
B.5 Dimethyl ether
 |
Fig. B.17 As in Fig. B.1, 32 brightest lines of the dimethyl ether, CH3 OCH3. Like in Fig. B.8, the asterisks next to some of the upper energy values indicate features where multiple transitions of the species contribute to the observed line. |
 |
Fig. B.18 As in Fig. B.1, 24 brightest lines of the 13C isotopologueof dimethyl ether, 13CH3OCH3 (see also Fig. B.17). |
 |
Fig. B.19 As in Fig. B.1, 24 + 8 brightest lines of the deuterated isotopologues of dimethyl ether: the antisymmetric form in the upper panels and lower left panel and the (rarer) symmetric form in the lower right panel (see also Fig. B.17). |
B.6 Acetaldehyde
 |
Fig. B.20 As in Fig. B.1, 24 brightest lines of one of the 13C isotopologues of acetaldehyde, 13CH3CHO, as expected from the synthetic spectrum. |
 |
Fig. B.21 As in Fig. B.1, 24 brightest lines of the second 13C isotopologueof acetaldehyde, CH3 13CHO, as expected from the synthetic spectrum. |
 |
Fig. B.22 As in Fig. B.1, 21 brightest lines of the deuterated isotopologue of acetaldehyde, CH3 CDO, as expected from the synthetic spectrum. |
B.7 Formic acid
 |
Fig. B.23 As in Fig. B.1, 24 brightest lines of formid acid, t-HCOOH, as expectedfrom the synthetic spectrum. See comment about overpredicted line at 338.20 GHz in 257 K panel in Sect. A.8. |
 |
Fig. B.24 As in Fig. B.1, 24 brightest lines of the second 13C isotopologueof formic acid, t-H13COOH, as expected from the synthetic spectrum. |
 |
Fig. B.25 As in Fig. B.1, deuterated isotopologue of formic acid, t-HCOOD, as expected from the synthetic spectrum. |
 |
Fig. B.26 As in Fig. B.1, 24 brightest lines of the deuterated isotopologue of formic acid, t-DCOOH, as expected from the synthetic spectrum. |
References
- Anderson, T., Crownover, R. L., Herbst, E., & De Lucia, F. C. 1988, ApJS, 67, 135 [NASA ADS] [CrossRef] [Google Scholar]
- Ball, J. A., Gottlieb, C. A., Lilley, A. E., & Radford, H. E. 1970, ApJ, 162, L203 [NASA ADS] [CrossRef] [Google Scholar]
- Baskakov, O. I. 1996, J. Mol. Spectr., 180, 266 [NASA ADS] [CrossRef] [Google Scholar]
- Baskakov, O. I., Bürger, H., & Jerzembeck, W. 1999, J. Mol. Spectr., 193, 33 [NASA ADS] [CrossRef] [Google Scholar]
- Baskakov, O. I., Lohilahti, J., & Horneman, V.-M. 2003, J. Mol. Spectr., 219, 191 [NASA ADS] [CrossRef] [Google Scholar]
- Baskakov, O. I., Alekseev, E. A., Motiyenko, R. A., et al. 2006, J. Mol. Spectr., 240, 188 [NASA ADS] [CrossRef] [Google Scholar]
- Belloche, A., Müller, H. S. P., Menten, K. M., Schilke, P., & Comito, C. 2013, A&A, 559, A47 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Belloche, A., Müller, H. S. P., Garrod, R. T., & Menten, K. M. 2016, A&A, 587, A91 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Belov, S. P., Tretyakov, M. Y., Kleiner, I., & Hougen, J. T. 1993, J. Mol. Spectr., 160, 61 [NASA ADS] [CrossRef] [Google Scholar]
- Bisschop, S. E., Jørgensen, J. K., van Dishoeck, E. F., & de Wachter, E. B. M. 2007, A&A, 465, 913 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bisschop, S. E., Schilke, P., Wyrowski, F., et al. 2013, A&A, 552, A122 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bizzocchi, L., Caselli, P., Spezzano, S., & Leonardo, E. 2014, A&A, 569, A27 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bouchez, A., Walters, A., Müller, H. S. P., et al. 2012, J. Quant. Spectr. Rad. Transf., 113, 1148 [NASA ADS] [CrossRef] [Google Scholar]
- Brown, R. D., Crofts, J. G., Godfrey, P. D., et al. 1975, ApJ, 197, L29 [NASA ADS] [CrossRef] [Google Scholar]
- Brown, R. D., Godfrey, P. D., McNaughton, D., Pierlot, A. P., & Taylor, W. H. 1990, J. Mol. Spectr., 140, 340 [NASA ADS] [CrossRef] [Google Scholar]
- Brünken, S., Sipilä, O., Chambers, E. T., et al. 2014, Nature, 516, 219 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Calcutt, H., Jørgensen, J. K., Müller, H. S. P., et al. 2018, A&A, 616, A90 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Carvajal, M., Kleiner, I., & Demaison, J. 2010, ApJS, 190, 315 [NASA ADS] [CrossRef] [Google Scholar]
- Cazaux, S., Tielens, A. G. G. M., Ceccarelli, C., et al. 2003, ApJ, 593, L51 [NASA ADS] [CrossRef] [Google Scholar]
- Cazaux, S., Caselli, P., & Spaans, M. 2011, ApJ, 741, L34 [NASA ADS] [CrossRef] [Google Scholar]
- Ceccarelli, C., Loinard, L., Castets, A., et al. 2001, A&A, 372, 998 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cernicharo, J., Kisiel, Z., Tercero, B., et al. 2016, A&A, 587, L4 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Charnley, S. B. 1997, ApJ, 481, 396 [NASA ADS] [CrossRef] [Google Scholar]
- Charnley, S. B., Tielens, A. G. G. M., & Millar, T. J. 1992, ApJ, 399, L71 [NASA ADS] [CrossRef] [Google Scholar]
- Charnley, S. B., Ehrenfreund, P., Millar, T. J., et al. 2004, MNRAS, 347, 157 [NASA ADS] [CrossRef] [Google Scholar]
- Chuang, K.-J., Fedoseev, G., Qasim, D., et al. 2017, MNRAS, 467, 2552 [NASA ADS] [Google Scholar]
- Churchwell, E., & Winnewisser, G. 1975, A&A, 45, 229 [NASA ADS] [Google Scholar]
- Comito, C., Schilke, P., Phillips, T. G., et al. 2005, ApJS, 156, 127 [NASA ADS] [CrossRef] [Google Scholar]
- Coudert, L. H., Drouin, B. J., Tercero, B., et al. 2013, ApJ, 779, 119 [NASA ADS] [CrossRef] [Google Scholar]
- Coutens, A., Vastel, C., Caux, E., et al. 2012, A&A, 539, A132 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Coutens, A., Jørgensen, J. K., van der Wiel, M. H. D., et al. 2016, A&A, 590, L6 [Google Scholar]
- Coutens, A., Willis, E. R., Garrod, R. T., et al. 2018, A&A, 612, A107 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Crockett, N. R., Bergin, E. A., Neill, J. L., et al. 2014, ApJ, 787, 112 [NASA ADS] [CrossRef] [Google Scholar]
- Cuadrado, S., Goicoechea, J. R., Roncero, O., et al. 2016, A&A, 596, L1 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cuppen, H. M., van Dishoeck, E. F., Herbst, E., & Tielens, A. G. G. M. 2009, A&A, 508, 275 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Demyk, K., Bottinelli, S., Caux, E., et al. 2010, A&A, 517, A17 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Drozdovskaya,M. N., Walsh, C., Visser, R., Harsono, D., & van Dishoeck, E. F. 2015, MNRAS, 451, 3836 [NASA ADS] [CrossRef] [Google Scholar]
- Duan, Y.-B., Ozier, I., Tsunekawa, S., & Takagi, K. 2003, J. Mol. Spectr., 218, 95 [NASA ADS] [CrossRef] [Google Scholar]
- Durig, J., Bucy, W. E., Wurrey, C. J., & Carreira, L. A. 1975, J. Phys. Chem., 79, 988 [Google Scholar]
- Elkeurti, M., Coudert, L. H., Medvedev, I. R., et al. 2010, J. Mol. Spectr., 263, 145 [NASA ADS] [CrossRef] [Google Scholar]
- Endres, C. P., Drouin, B. J., Pearson, J. C., et al. 2009, A&A, 504, 635 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Faure, A., Faure, M., Theulé, P., Quirico, E., & Schmitt, B. 2015, A&A, 584, A98 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fedoseev, G., Cuppen, H. M., Ioppolo, S., Lamberts, T., & Linnartz, H. 2015, MNRAS, 448, 1288 [NASA ADS] [CrossRef] [Google Scholar]
- Fedoseev, G., Chuang, K.-J., van Dishoeck, E. F., Ioppolo, S., & Linnartz, H. 2016, MNRAS, 460, 4297 [NASA ADS] [CrossRef] [Google Scholar]
- Fisher, J., Paciga, G., Xu, L.-H., et al. 2007, J. Mol. Spectr., 245, 7 [NASA ADS] [CrossRef] [Google Scholar]
- Fourikis, N., Sinclair, M. W., Robinson, B. J., Godfrey, P. D., & Brown, R. D. 1974, Aust. J. Phys., 27, 425 [NASA ADS] [CrossRef] [Google Scholar]
- Fuchs, G. W., Cuppen, H. M., Ioppolo, S., et al. 2009, A&A, 505, 629 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Furuya, K., Aikawa, Y., Sakai, N., & Yamamoto, S. 2011, ApJ, 731, 38 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Furuya, K., van Dishoeck, E. F., & Aikawa, Y. 2016, A&A, 586, A127 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Gardner, F. F., Whiteoak, J. B., Reynolds, J., Peters, W. L., & Kuiper, T. B. H. 1989, MNRAS, 240, 35P [NASA ADS] [CrossRef] [Google Scholar]
- Garrod, R. T. 2013, ApJ, 765, 60 [Google Scholar]
- Garrod, R. T., Weaver, S. L. W., & Herbst, E. 2008, ApJ, 682, 283 [NASA ADS] [CrossRef] [Google Scholar]
- Gottlieb, C. A. 1973, in Molecules in the Galactic Environment, eds. M. A. Gordon & L. E. Snyder, 181 [Google Scholar]
- Gottlieb, C. A., Ball, J. A., Gottlieb, E. W., & Dickinson, D. F. 1979, ApJ, 227, 422 [NASA ADS] [CrossRef] [Google Scholar]
- Goumans, T. P. M., Uppal, M. A., & Brown, W. A. 2008, MNRAS, 384, 1158 [NASA ADS] [CrossRef] [Google Scholar]
- Groner, P., & Durig, J. R. 1977, J. Chem. Phys., 66, 1856 [NASA ADS] [CrossRef] [Google Scholar]
- Groner, P., Albert, S., Herbst, E., & De Lucia F. C. 1998, ApJ, 500, 1059 [NASA ADS] [CrossRef] [Google Scholar]
- Guarnieri, A. 2005, Z. Nat. Teil A, 60, 619 [Google Scholar]
- Guarnieri, A., & Huckauf, A. 2003, Z. Nat. Teil A, 58, 275 [Google Scholar]
- Harju, J., Sipilä, O., Brünken, S., et al. 2017, ApJ, 840, 63 [NASA ADS] [CrossRef] [Google Scholar]
- Hasegawa, T. I., & Herbst, E. 1993, MNRAS, 263, 589 [NASA ADS] [CrossRef] [Google Scholar]
- Ikeda, M., Duan, Y.-B., Tsunekawa, S., & Takagi, K. 1998, ApJS, 117, 249 [NASA ADS] [CrossRef] [Google Scholar]
- Ilyushin, V., Kryvda, A., & Alekseev, E. 2009, J. Mol. Spectr., 255, 32 [NASA ADS] [CrossRef] [Google Scholar]
- Ioppolo, S., Cuppen, H. M., van Dishoeck, E. F., & Linnartz, H. 2011, MNRAS, 410, 1089 [NASA ADS] [CrossRef] [Google Scholar]
- Irvine, W. M., Friberg, P., Kaifu, N., et al. 1990, A&A, 229, L9 [NASA ADS] [Google Scholar]
- Isokoski, K., Bottinelli, S., & van Dishoeck, E. F. 2013, A&A, 554, A100 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jaber, A. A., Ceccarelli, C., Kahane, C., & Caux, E. 2014, ApJ, 791, 29 [Google Scholar]
- Jacq, T., Walmsley, C. M., Mauersberger, R., et al. 1993, A&A, 271, 276 [NASA ADS] [Google Scholar]
- Jørgensen, J. K., Bourke, T. L., Nguyen Luong, Q., & Takakuwa, S. 2011, A&A, 534, A100 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jørgensen, J. K., Favre, C., Bisschop, S. E., et al. 2012, ApJ, 757, L4 [NASA ADS] [CrossRef] [Google Scholar]
- Jørgensen, J. K., van der Wiel, M. H. D., Coutens, A., et al. 2016, A&A, 595, A117 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Kleiner, I., Lovas, F. J., & Godefroid, M. 1996, J. Phys. Chem. Ref. Data, 25, 1113 [CrossRef] [Google Scholar]
- Koerber, M., Bisschop, S. E., Endres, C. P., et al. 2013, A&A, 558, A112 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Langer, W. D., Graedel, T. E., Frerking, M. A., & Armentrout, P. B. 1984, ApJ, 277, 581 [NASA ADS] [CrossRef] [Google Scholar]
- Lattanzi, V., Walters, A., Drouin, B. J., & Pearson, J. C. 2008, ApJS, 176, 536 [NASA ADS] [CrossRef] [Google Scholar]
- Lauvergnat, D., Coudert, L. H., Klee, S., & Smirnov, M. 2009, J. Mol. Spectr., 256, 204 [NASA ADS] [CrossRef] [Google Scholar]
- Ligterink, N. F. W., Coutens, A., Kofman, V., et al. 2017, MNRAS, 469, 2219 [NASA ADS] [CrossRef] [Google Scholar]
- Ligterink, N., Calcutt, H., Coutens, A., et al. 2018, A&A, 619, A28 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Linsky, J. L. 2003, Space Sci. Rev., 106, 49 [NASA ADS] [CrossRef] [Google Scholar]
- Lykke, J. M., Coutens, A., Jørgensen, J. K., et al. 2017, A&A, 597, A53 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Maeda, A., De Lucia, F. C., & Herbst, E. 2008a, J. Mol. Spectr., 251, 293 [NASA ADS] [CrossRef] [Google Scholar]
- Maeda, A., Medvedev, I. R., De Lucia, F. C., Herbst, E., & Groner, P. 2008b, ApJS, 175, 138 [NASA ADS] [CrossRef] [Google Scholar]
- Margulès, L., Coudert, L. H., Møllendal, H., et al. 2009, J. Mol. Spectr., 254, 55 [NASA ADS] [CrossRef] [Google Scholar]
- Margulès, L., Motiyenko, R. A., Ilyushin, V. V., & Guillemin, J. C. 2015, A&A, 579, A46 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Milam, S. N., Savage, C., Brewster, M. A., Ziurys, L. M., & Wyckoff, S. 2005, ApJ, 634, 1126 [NASA ADS] [CrossRef] [Google Scholar]
- Müller, H. S. P., Thorwirth, S., Roth, D. A., & Winnewisser, G. 2001, A&A, 370, L49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Müller, H. S. P., Schlöder, F., Stutzki, J., & Winnewisser, G. 2005, J. Mol. Struct., 742, 215 [NASA ADS] [CrossRef] [Google Scholar]
- Müller, H. S. P., Belloche, A., Xu, L.-H., et al. 2016, A&A, 587, A92 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Neill, J. L., Crockett, N. R., Bergin, E. A., Pearson, J. C., & Xu, L.-H. 2013, ApJ, 777, 85 [NASA ADS] [CrossRef] [Google Scholar]
- Neill, J. L., Bergin, E. A., Lis, D. C., et al. 2014, ApJ, 789, 8 [NASA ADS] [CrossRef] [Google Scholar]
- Öberg, K. I., Garrod, R. T., van Dishoeck, E. F., & Linnartz, H. 2009, A&A, 504, 891 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Oesterling, L. C., Ferguson, D. W., Herbst, E., & De Lucia F. C. 1995, J. Mol. Spectr., 172, 469 [NASA ADS] [CrossRef] [Google Scholar]
- Oesterling, L. C., Albert, S., De Lucia, F. C., Sastry, K. V. L. N., & Herbst, E. 1999, ApJ, 521, 255 [NASA ADS] [CrossRef] [Google Scholar]
- Parise, B., Ceccarelli, C., Tielens, A. G. G. M., et al. 2002, A&A, 393, L49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Parise, B., Castets, A., Herbst, E., et al. 2004, A&A, 416, 159 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Parise, B., Ceccarelli, C., Tielens, A. G. G. M., et al. 2006, A&A, 453, 949 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pearson, J. C., Sastry, K. V. L. N., Winnewisser, M., Herbst, E., & De Lucia, F. C. 1995, J. Phys. Chem. Ref. Data, 24, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Pearson, J. C., Sastry, K. V. L. N., Herbst, E., & De Lucia, F. C. 1996, J. Mol. Spectr., 175, 246 [NASA ADS] [CrossRef] [Google Scholar]
- Pearson, J. C., Brauer, C. S., & Drouin, B. J. 2008, J. Mol. Spectr., 251, 394 [NASA ADS] [CrossRef] [Google Scholar]
- Pearson, J. C., Yu, S., & Drouin, B. J. 2012, J. Mol. Spectr., 280, 119 [NASA ADS] [CrossRef] [Google Scholar]
- Peng, T.-C., Despois, D., Brouillet, N., Parise, B., & Baudry, A. 2012, A&A, 543, A152 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Perrin, A., Flaud, J.-M., Bakri, B., et al. 2002, J. Mol. Spectr., 216, 203 [NASA ADS] [CrossRef] [Google Scholar]
- Persson, M. V., Jørgensen, J. K., & van Dishoeck, E. F. 2013, A&A, 549, L3 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Persson, M. V., Harsono, D., Tobin, J. J., et al. 2016, A&A, 590, A33 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Persson, M. V., Jørgensen, J. K., Müller, H. S. P., et al. 2018, A&A, 610, A54 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pickett, H. M., Poynter, I. R. L., Cohen, E. A., et al. 1998, J. Quant. Spectr., 60, 883 [Google Scholar]
- Plummer, G. M., Herbst, E., De Lucia, F., & Blake, G. A. 1984, ApJS, 55, 633 [NASA ADS] [CrossRef] [Google Scholar]
- Prodanović, T., Steigman, G., & Fields, B. D. 2010, MNRAS, 406, 1108 [NASA ADS] [Google Scholar]
- Richard, C., Margulès, L., Caux, E., et al. 2013, A&A, 552, A117 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Schöier, F. L., Jørgensen, J. K., van Dishoeck, E. F., & Blake, G. A. 2002, A&A, 390, 1001 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Smith, R. L., Pontoppidan, K. M., Young, E. D., & Morris, M. R. 2015, ApJ, 813, 120 [NASA ADS] [CrossRef] [Google Scholar]
- Snyder, L. E., Buhl, D., Schwartz, P. R., et al. 1974, ApJ, 191, L79 [NASA ADS] [CrossRef] [Google Scholar]
- Taquet, V., López-Sepulcre, A., Ceccarelli, C., et al. 2013, ApJ, 768, L29 [NASA ADS] [CrossRef] [Google Scholar]
- Taquet, V., Charnley, S. B., & Sipilä, O. 2014, ApJ, 791, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Taquet, V., Wirström, E. S., Charnley, S. B., et al. 2017, A&A, 607, A20 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Tercero, B., Margulès, L., Carvajal, M., et al. 2012, A&A, 538, A119 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Tielens, A. G. G. M. 1983, A&A, 119, 177 [NASA ADS] [Google Scholar]
- Tielens, A. G. G. M., & Hagen, W. 1982, A&A, 114, 245 [Google Scholar]
- Turner, B. E. 1977, ApJ, 213, L75 [NASA ADS] [CrossRef] [Google Scholar]
- van Dishoeck, E. F., Blake, G. A., Jansen, D. J., & Groesbeck, T. D. 1995, ApJ, 447, 760 [NASA ADS] [CrossRef] [Google Scholar]
- Walters, A., Schäfer, M., Ordu, M. H., et al. 2015, J. Mol. Spectr., 314, 6 [NASA ADS] [CrossRef] [Google Scholar]
- Watanabe, N., & Kouchi, A. 2002, ApJ, 571, L173 [NASA ADS] [CrossRef] [Google Scholar]
- Willaert, F., Møllendal, H., Alekseev, E., et al. 2006, J. Mol. Struct., 795, 4 [NASA ADS] [CrossRef] [Google Scholar]
- Wilson, T. L., & Rood, R. 1994, ARA&A, 32, 191 [NASA ADS] [CrossRef] [Google Scholar]
- Winnewisser, G., & Churchwell, E. 1975, ApJ, 200, L33 [NASA ADS] [CrossRef] [Google Scholar]
- Winnewisser, M., Winnewisser, B. P., Stein, M., et al. 2002a, J. Mol. Spectr., 216, 259 [NASA ADS] [CrossRef] [Google Scholar]
- Winnewisser, M., Winnewisser, B. P., Stein, M., et al. 2002b, J. Mol. Spectr., 216, 259 [NASA ADS] [CrossRef] [Google Scholar]
- Zernickel, A., Schilke, P., Schmiedeke, A., et al. 2012, A&A, 546, A87 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Zuckerman, B., Ball, J. A., & Gottlieb, C. A. 1971, ApJ, 163, L41 [NASA ADS] [CrossRef] [Google Scholar]
- Zuckerman, B., Turner, B. E., Johnson, D. R., et al. 1975, ApJ, 196, L99 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
Derived column densities for the isotopologues of the complex organics and abundances relative to the main isotopologue at the position offset by 0.5″ (60 AU) from IRAS16293B.
Division of molecules into “hot” and “cold” from the interferometric observations of IRAS16293 (this study) and high-mass hot cores (Bisschop et al. 2007), an indication of whether or not the species are detected in the single-dish study of IRAS16293 (Jaber et al. 2014) and the binding energies of each species (from the tabulation by Garrod 2013).
Inferred abundances and deuterium fractionation (from interferometric and single-dish data, respectively) for different organic molecules.
All Figures
|
Fig. 1 Rotation diagrams for well-isolated lines of CH3OD (upper panel) and CH3CHDOH (lower panel). For the former, both rotation temperature and column density can be constrained, while the limited range of CH3 OD transitions only allow us to constrain the column density for that species. In both panels, the best fit is shown with the solid line with our conservative estimates on the uncertainties represented by the dotted lines (the best fit values given in the upper right corner of each panels). The dashed lines in both panels indicate the best fits assuming a fixed rotation temperature of 125 K: this temperature clearly does not match the data for CH3 OD over the range of upper energies for the observed lines. |
| In the text | |
|
Fig. 2 Representative maps for lines of CH |
| In the text | |
|
Fig. 3 Spectrum of optically thick transitions of CH2DOH at 356.95 GHz toward the half-beam (upper) and full-beam (lower) offset positions from IRAS16293B. In the lower panel, synthetic spectra for CH2DOH are overlaid:those are both calculated for the same column density (Table 1) but with temperatures of 50 K (blue) and 300 K (red). |
| In the text | |
|
Fig. 4 Schematic representation of the results from this paper: the discussed molecules are grouped according to their D/H ratios and excitation temperatures. The species underlined can be shown to have lower 12C/13C ratios by a factor 2 than typical values for the local ISM (although only tentatively for methyl formate and ethanol), while the remainder are either consistent with a standard ISM ratio or have too optically thick lines of the main isotopologuefor a reliable independent estimate. |
| In the text | |
|
Fig. 5 Comparison between the D/H ratios (per H-atom) for the complex organics sorted by species (left panel) and the leading radical in the formation (right panel). For each species, the PILS observations are represented by the first, red, bar and the predictions in the models by Taquet et al. (2014) after 1.1 × 105, 1.5 × 105 and 2.0 × 105 years by the second (blue), third (orange) and fourth (green) bars, respectively (see also legend in the part of the right panel). For the model at 2.0 × 105 years, the D/H ratios have been multiplied by a factor of 10 to enable comparisons. |
| In the text | |
|
Fig. B.1 The15+16 brightest lines of CH3OD (a-type in the upper panels and e-type in the lower panels) toward the full-beam offset position. The lines are sorted according to Eup given in K in the upper left corner of each panel. The windows are centered on these specific lines. |
| In the text | |
|
Fig. B.2 As in Fig. B.1, zoomed-in to highlight the fits. Overplotted are the best fit models using excitation temperatures 20% above (dotted) and 20% (dashed) below the best fit value. |
| In the text | |
|
Fig. B.3 As in Fig. B.1, zoomed-in to highlight the fits. Overplotted are the best fit models using column densities 20% above (dotted) and 20% (dashed) below the best fit value. |
| In the text | |
|
Fig. B.4 The24 brightest optically thin lines of CH2DOH as expected from the synthetic spectrum toward the full-beam offset position. In the cases, where other lines fall in the band that are strongly optically thick at 50 or 300 K, they have been marked with a plus-sign or cross, respectively. The discrepancies for the lines at 349.62 and 346.82 GHz likely reflect issues with the catalogue entries for these high excitation transitions. |
| In the text | |
|
Fig. B.5 As in Fig. B.1, 21 brightest lines of CH |
| In the text | |
|
Fig. B.6 As in Fig. B.1, 32 brightest lines of ethanol as expected from the synthetic spectrum toward the full-beam offset position. |
| In the text | |
|
Fig. B.7 As in Fig. B.1, 16 brightest lines of a-13CH3CH2OH (upper panels) and a-CH |
| In the text | |
|
Fig. B.8 As in Fig. B.1, 16 brightest lines of a-CH3CHDOH toward the half-beam (upper panel) and full-beam (lower panel) offset positions. The asterisks next to the upper energy level in some panels indicate features for which the multiple transitions (from the same species) overlap. |
| In the text | |
|
Fig. B.9 As in Fig. B.1, a-CH3CH2OD the upper panels show the spectra toward the half-beam position and the lower panels the full-beam (lower) offset position. |
| In the text | |
 |
Fig. B.12 As in Fig. B.1, 24 brightest lines of CH3OCHO as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.13 As in Fig. B.1, 16 + 16 brightest lines of deuterated methyl formate: DCOOCH3 in the upper panels and CH2DOCHO in the lower panels. |
| In the text | |
|
Fig. B.14 As in Fig. B.1, 16 brightest lines of 13C methyl formate, CH3O13CHO toward the half-beam (upper panel) and full-beam (lower panel) offset positions. |
| In the text | |
|
Fig. B.15 As in Fig. B.1, the brightest lines of the 13C-isotopologues of ketene with 13CH2CO shown in the upper panels and CH213CO in the lower panels. |
| In the text | |
|
Fig. B.16 As in Fig. B.1, 16 brightest lines of the singly deuterated-isotopologues of ketene CHDCO (upper panels). The lower panels show the eight brightest lines of doubly deuterated ketene, CD2 CO (left panel) and the 18O isotopologueCH2C18O (right panel). For those we assume the same D/H ratio as for the singly deuterated species represents an upper limit/tentative identification. |
| In the text | |
|
Fig. B.17 As in Fig. B.1, 32 brightest lines of the dimethyl ether, CH3 OCH3. Like in Fig. B.8, the asterisks next to some of the upper energy values indicate features where multiple transitions of the species contribute to the observed line. |
| In the text | |
|
Fig. B.18 As in Fig. B.1, 24 brightest lines of the 13C isotopologueof dimethyl ether, 13CH3OCH3 (see also Fig. B.17). |
| In the text | |
|
Fig. B.19 As in Fig. B.1, 24 + 8 brightest lines of the deuterated isotopologues of dimethyl ether: the antisymmetric form in the upper panels and lower left panel and the (rarer) symmetric form in the lower right panel (see also Fig. B.17). |
| In the text | |
|
Fig. B.20 As in Fig. B.1, 24 brightest lines of one of the 13C isotopologues of acetaldehyde, 13CH3CHO, as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.21 As in Fig. B.1, 24 brightest lines of the second 13C isotopologueof acetaldehyde, CH3 13CHO, as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.22 As in Fig. B.1, 21 brightest lines of the deuterated isotopologue of acetaldehyde, CH3 CDO, as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.23 As in Fig. B.1, 24 brightest lines of formid acid, t-HCOOH, as expectedfrom the synthetic spectrum. See comment about overpredicted line at 338.20 GHz in 257 K panel in Sect. A.8. |
| In the text | |
|
Fig. B.24 As in Fig. B.1, 24 brightest lines of the second 13C isotopologueof formic acid, t-H13COOH, as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.25 As in Fig. B.1, deuterated isotopologue of formic acid, t-HCOOD, as expected from the synthetic spectrum. |
| In the text | |
|
Fig. B.26 As in Fig. B.1, 24 brightest lines of the deuterated isotopologue of formic acid, t-DCOOH, as expected from the synthetic spectrum. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.