| Issue |
A&A
Volume 694, February 2025
|
|
|---|---|---|
| Article Number | A299 | |
| Number of page(s) | 15 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202451990 | |
| Published online | 20 February 2025 | |
Hydrogenation of acetaldehyde on interstellar ice analogs results in limited destruction
1
Departamento de Astrofísica Molecular, Instituto de Física Fundamental (IFF-CSIC),
C/ Serrano 121,
28006
Madrid,
Spain
2
Institute of Low Temperature Science, Hokkaido University N19W8,
Kita-ku,
Sapporo,
Hokkaido
060-0819
Japan
★ Corresponding authors; This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
26
August
2024
Accepted:
23
December
2024
Abstract
Context. Acetaldehyde (CH3CHO) is one of the most abundant interstellar complex organic molecules and its hydrogenation has important implications in several fundamental processes of interstellar chemistry, such as deuterium fractionation, reactive desorption, or the relation between organic functional groups of detected molecules.
Aims. We seek to determine what the main hydrogenation paths of CH3CHO are. As a partially unsaturated molecule, CH3CHO can have links with more hydrogenated species, such as ethanol (C2H5OH), or with more unsaturated ones, such as ketene (H2CCO).
Methods. We used highly accurate quantum chemical calculations to determine the reaction rate constants for the CH3CHO + H/D reaction. We later studied, using more approximated methods, the fate of the majoritarian product of the reaction, the acetyl radical CH3CO after subsequent reaction with hydrogen or deuterium atoms. Our theoretical results were tested with our experiments on the hydrogenation and deuteration of CH3CHO ice.
Results. We find that acetaldehyde resists hydrogenation, with only a 10% of conversion to products different than CH3CHO. This is due to a predominance of H abstraction at the HCO moiety, with reaction rate constants up to four orders of magnitude higher than the next possible reaction channel, which is hydrogenation at the aldehydic carbon. The formed CH3CO radical experiences barrierless or nearly barrierless reactions in all possible reaction positions, reforming CH3CHO and creating a closed loop that protects the molecule against hydrogenation. We constrained the branching ratios for the second reaction from the experiments. Our experiments agree with the calculations and from the combination of both we can explain the presence of H2CCO, CO, CH4, C2H5OH, H2CO, or CH3OH as minor products at the end of the reaction. We provide recommendations for future modeling efforts.
Conclusions. Our results show limited destruction of acetaldehyde, reinforcing the vision of this molecule as an abundant and resilient COM. From the experiments, we are not able to observe the reactive desorption of this molecule. Our results align with other modeling works, showing that the link between CH3CHO and C2H5OH is not direct. Finally, our results can explain the excess of CH3CDO found in prestellar cores.
Key words: astrochemistry / molecular data / methods: laboratory: molecular / methods: numerical / ISM: molecules
© The Authors 2025
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
The emergence of chemical complexity in the interstellar medium (ISM) is one of the most fascinating topics in astrochemistry. Although the concept of “complexity” is generally ill-defined, a molecule composed of six or more atoms, with at least one that is carbon, is typically considered a complex organic interstellar molecule (Herbst & Van Dishoeck 2009). Given the harsh conditions in the ISM, the formation mechanisms of these molecules are vastly different from those on Earth. In the ISM, particularly within the ice-covered dust grains that account for approximately 1% of its mass (Bohlin et al. 1978), many molecules are formed through radical chemistry.
Significant effort has been devoted to studying the synthesis of complex organic molecules (COMs) on interstellar ices (Watanabe & Kouchi 2002; Fuchs et al. 2009; Rimola et al. 2014; Garrod 2013; Simons et al. 2020; Jin & Garrod 2020; Garrod et al. 2022; Enrique-Romero et al. 2022; Ferrero et al. 2023; Molpeceres et al. 2024a, to give a few representative examples). Although the processing reactions of COMs, those that convert or destroy already-formed COMs, have also garnered attention, they remain comparatively underexplored.
Examples in the literature include the systematic study of COM hydrogenation (Alvarez-Barcia et al. 2018), the hydrogenation of heterocycles (Miksch et al. 2021), and the processing of formic acid and thioformic acid (Molpeceres et al. 2022a). However, the chemical processing of COMs on interstellar grains has important implications for the evolution of chemical complexity. Among these, hydrogen abstraction (H-abstraction) reactions play a pivotal role. These reactions involve a radical (e.g., H, OH, NH2) abstracting a hydrogen atom from a COM, producing a closed-shell hydrogenated molecule (e.g., H2, H2O, NH3) and leaving behind a parent radical of the COM. For instance, we recently demonstrated that for hydroxylamine (NH2OH), H-abstraction reactions, followed by O or N addition, can account for the apparent disappearance of this molecule in the ISM (Molpeceres et al. 2023). Besides, following similar arguments, deuterium enrichment of molecules and COMs (see, e.g., the review of Ceccarelli et al. 2014), is enhanced through H-abstraction reactions, following a cycle of H abstraction and D addition (Molpeceres et al. 2022a; Nguyen et al. 2021a; Lamberts & Kästner 2017b). Furthermore, the same cycle can fuel the process of reactive desorption to return the ice-synthesized molecules to the gas phase. This is due to the increase in reactive events for a fixed probability of chemical desorption per reaction (Garrod et al. 2007), as was evinced by chemical desorption experiments by some of us (Oba et al. 2018; Nguyen et al. 2020) on H2S or PH3. Clearly, H-abstraction reactions are critical components of modern astrochemical reaction networks and warrant more detailed investigation.
With advancements in astrochemistry, the pool of molecules requiring detailed study is growing not only in number but also in complexity. For instance, considering the examples of H2S and PH3 mentioned above, these molecules have relatively simple reaction networks (Lamberts & Kästner 2017b; Molpeceres & Kästner 2021; Nguyen et al. 2021a). Starting from their atomic precursors (S and P), the fully hydrogenated molecule is the most stable product. Additionally, H-abstraction reactions exhibit increasing activation energies, while H addition remains barrierless. These traits make such molecules easier to detect in experimental setups, as the reaction products are limited to one stable product and undetectable reactive radicals. However, the complexity increases significantly when a carbon atom is introduced, as in carbon monosulfide (CS). Adding carbon leads to reaction networks with approximately 15 reactions en route to forming CH3SH (Lamberts 2018; Nguyen et al. 2023), including irreversible pathways such as CH3 + H2S. This highlights the intricate nature of hydrogenation sequences. Studying these networks for COMs routinely identified in cold environments (Bacmann et al. 2012; Bacmann & Faure 2014; Jiménez-Serra et al. 2021; Jiménez-Serra et al. 2016; Megías et al. 2022; Scibelli et al. 2021) becomes increasingly challenging, whether approached experimentally or theoretically.
The molecule examined in this paper, acetaldehyde (CH3CHO), is one of the most abundant and extensively studied COMs that can form on ice grains (Jin & Garrod 2020; Lamberts et al. 2019; Ferrero et al. 2023; Molpeceres et al. 2024a), albeit with some challenges (Enrique-Romero et al. 2021; Perrero et al. 2023). Acetaldehyde can also be synthesized in the gas phase (Vazart et al. 2020). Although CH3CHO qualifies as a COM, its reaction with hydrogen remains experimentally tractable (Bisschop et al. 2007), yet interpreting the experimental results is far from straightforward. Hydrogenation of CH3CHO reportedly produces a mixture of acetaldehyde (CH3CHO), ethanol (C2 H5 OH), methane (CH4), carbon monoxide (CO), formaldehyde (H2CO), and methanol (CH3OH). However, the latter three products are linked through a sequential hydrogenation pathway (Watanabe & Kouchi 2002). Meanwhile, CH4 and C2H5OH could, in principle, arise from alternative pathways rather than the direct hydrogenation of CH3CHO. This complexity underscores the need to better understand the precise mechanism of CH3CHO hydrogenation1. Such a detailed understanding is essential for proposing potential deuteration pathways and assessing the likelihood of reactive desorption, as was discussed earlier.
A mechanistic understanding of this reaction can only be achieved through a synergistic combination of accurate theoretical calculations and experimental work. This is the approach we have adopted in this study paper, in which we integrate quantum chemical calculations of the hydrogenation of CH3CHO, incorporating a rigorous treatment of nuclear quantum effects, with tailored experiments on the CH3CHO + H reaction. Furthermore, our work aims to unify the existing knowledge on this reaction and related reactions, such as CO + H and H2CCO + H (Watanabe & Kouchi 2002; Fuchs et al. 2009; Mondal et al. 2021; Fedoseev et al. 2022; Ibrahim et al. 2022; Ferrero et al. 2023), by resolving discrepancies where they exist or providing justified support where they align. The structure of this manuscript is as follows. In Sect. 2, we provide a concise overview of our methodology. Section 3 is dedicated to the presentation of our findings. In Sect. 4, we discuss the results, placing them in the context of experimental, modeling, and broader astrochemical perspectives. Finally, we conclude with key remarks in Sect. 5.
2 Methods
2.1 Computational details
Quantum chemical calculations were performed with a twofold objective. First, we used the results of a preliminary exploration to guide our experimental search. Second, when the full experimental procedure was completed, we expanded our quantum chemical calculations to explain the experiments, leading to a self-consistent explanation of CH3CHO chemistry under ISM conditions. We can categorize our simulations into three different categories. First, we carried out calibration calculations using a small two-water model to test the influence of explicit water molecules in the hydrogenation of CH3CHO (Appendix A). Second, we derived the instanton rate constants (Rommel et al. 2011; Kästner 2014) for H addition, H abstraction, and D addition on CH3CHO. Instanton theory is a unification of path integral theory and transition state theory to simulate the quantum nature of the nuclei in the reactant and transition states of a reaction. An instanton represents the most probable tunneling path, which in our formulation is discretized in a number of points or beads, and optimized to a first-order saddle point, like in conventional transition state theory. In this case, an optimization of the tunneling path with a maximum transition probability (or minimum Euclidean action) was carried out. Third and last, we investigated additional reactions with hydrogen in the acetyl radical (CH3CO), the preferred product of the first reaction with H (see Sect. 3). Our quantum chemical calculations are designed to prioritize accuracy in the electronic structure calculations over cluster representativity. Acetaldehyde is shown to be a COM with a relatively weak binding with the water surface (Molpeceres et al. 2022b; Ferrero et al. 2022), which normally leads to a small influence of the water matrix on the reaction rate constants (see, for example Ferrero et al. 2023; Molpeceres et al. 2024b, for two recent examples). The instanton rate constants were calculated using a sequential cooling scheme starting from a temperature close to the crossover temperature defined as:
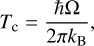 (1)
(1)
where Ω corresponds to the imaginary frequency of vibration at the transition state. Tc is an estimator of the temperature at which the tunneling effects in the rate constant start to dominate over the purely thermal ones (Gillan 1987). Because the determining quantity to compare with experiments is the reaction rate constant, which we obtained from accurate semiclassical instanton theory calculations, the inclusion of surface effects in our calculations was done via the implicit surface approach (Meisner et al. 2017); in other words, by making the rotational partition faction equal to one in the calculation of the reaction rate constant. Omitting the ice matrix allows us to use a significantly better level of theory than when including an ice matrix. In particular, the level of theory used in this work is the combination of the rev-DSD-PBEP86(D4)/jun-cc-pV(T+d)Z level (i.e., double hybrid functional with a large basis set) (Kozuch & Martin 2011; Santra et al. 2019; Dunning 1989; Papajak et al. 2011; Caldeweyher et al. 2019) for the molecular geometries and molecular Hessian and the (U)CCSD(T)/aug-cc-pVTZ (Bartlett & Purvis 1978; Purvis & Bartlett 1982; Dunning 1989; Knowles et al. 1993; Woon & Dunning 1994) gold standard method for correcting the electronic energies2. This level was used for the determination of stationary points in the respective potential energy surface (PES), calculation of the zero-point vibrational energies (ZPVEs), and the calculation of rate constants in the interaction of H with CH3CHO. To incorporate the effect of a polar environment – that is, H2O – we also computed single point energies using the conductor-like polarizable continuum (CPCM) (Truong & Stefanovich 1995; Barone & Cossi 1998; Garcia-Ratés & Neese 2020) model, employing a dielectric constant extrapolated for amorphous solid water (ASW) of є = 600, as has been used in recent astrochemical literature (Nguyen et al. 2019). We present models with and without CPCM correction. The hydrogenation of the CH3CO was investigated with rev-DSD-PBEP86(D4)/jun-cc-pV(T+d)Z; that is, without further coupled cluster refinement, although CPCM single points at the density functional theory (DFT) level are also reported. The last set of calculations was carried out using a broken-symmetry DFT formalism, first converging the triplet electronic state and then rotating the orbitals of the incoming reactant (H) to guarantee the correct spin state for the system. The rate constants for the reactions starting with the CH3CO as a reactant were obtained from classical transition state theory, including the tunneling correction from an asymmetric Eckart barrier (Eckart 1930). The energies coming from pure broken symmetry DFT calculations were subjected to a higher error than when correcting the electronic energies with a high-level method. However, the lack of a reliable single reference solution in the case of biradical species such as the ones involved in the CH3CO + H reaction prompted us to continue with this protocol. The agreement between experiments and theory found in this work serves as a proxy benchmark for the validity of our method in this particular case.
All our electronic structure calculations used the ORCA 5.0.4 code (Neese et al. 2020). Geometry optimizations and instanton calculations used a developer version of the DL-FIND code (Kästner et al. 2009) interfaced with CHEMSHELL (Metz et al. 2014)3.
2.2 Experiments
All experiments were performed using an experimental apparatus named Apparatus for Surface Reaction in Astrophysics (ASURA). The details of the ASURA system have been described in previous studies (Watanabe et al. 2006; Nagaoka et al. 2007; Nguyen et al. 2020, 2021b, 2023). In brief, it consists of an ultrahigh vacuum chamber with the basic pressure of 10−8 Pa; an aluminum (Al) substrate attached to a He cryostat, an atomic source, a quadrupole mass spectrometer (QMS), and a Fourier transform infrared spectrometer (FTIR). The surface temperature was regulated between 5 and 300 K.
The chemical reactions of solid acetaldehyde (CH3CHO) with H (or D) atoms were studied on both an Al substrate and compact amorphous solid water (c-ASW) at low temperatures (typically 10 K). The c-ASW was made by the vapor deposition of water onto the substrate maintained at 110 K, with an estimated thickness of ~20 monolayers (ML; 1 ML = 1 × 1015 molecules cm−2). The substrate was cooled down to 10 K after the c-ASW layer production. Gaseous CH3CHO was pre-deposited onto c-ASW with the deposition rate of 1 ML minute−1. The layer thickness of CH3CHO was adjusted to 1 ML, which was estimated using the peak area of the CO stretching band at 1728 cm−1 and its absorption coefficient of 8.0 × 10−18 cm molecule−1 (Bisschop et al. 2007). The pre-deposited CH3CHO layer was exposed to the H and D atoms produced via the dissociation of H2 (or D2) in a microwave-discharged plasma in the atomic source chamber. The formed H (D) atoms were cooled to 100 K by multiple collisions with the inner wall of the Al pipe at 100 K (Nagaoka et al. 2007). The flux of H and D atoms was estimated as 5.0 × 1014 and 3.4 × 1014 cm−2 s−1, respectively, following the method of Oba et al. (2014). During the process of H (or D) exposure on the sample solid, interactions between CH3CHO and H (D) atoms were observed in situ using FTIR with a resolution of 2 cm−1. The reactants and products desorbed from the surface were monitored by the QMS via the temperature programmed desorption (TPD) method, with a ramping rate of 4 K minute−1 .
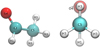 |
Fig. 1 Acetaldehyde molecule with the positions where the hydrogenation and deuteration reactions were sampled and Newman projection of the molecular model showing the considered CH3CHO syn conformer. |
3 Results
3.1 Theoretical exploration
3.1.1 Description of CH3CHO + H
The study of the H-abstraction and addition reactions necessary to rationalize our experiments begins with the evaluation of the energetics for all the possible reactions reaction channels for CH3CHO + H. The molecular structure of syn-CH3CHO and the atomic positions where the reaction can take place are depicted in Fig 1. For each reaction, we assume that energy dissipation occurs via the surface, treating the reactions presented in this study as elementary. The possible reactions are:
 (R1)
(R1)
 (R2)
(R2)
 (R3)
(R3)
 (R4)
(R4)
 (R5)
(R5)
The energetic parameters for the reactions, reaction energies (∆HR), activation energies (∆H‡), and imaginary frequencies of vibration at the TS (Ω) are gathered in Table 1. In the first place, we confirm that all possible reactions are exothermic reactions that proceed with a moderate ∆H‡, as is expected for closed- shell organic molecules. It is evident from the data in Table 1 that reaction (R5) is, under no circumstances, competitive with Reactions (R1)–(R4), due to the extremely high activation energies. This conclusion allows us to determine that all CH4 and its isotopologs are formed in the experiment from radical-radical recombinations of secondary products of the reaction (see Sect. 4.1). The other reactions present barriers between ~20 and 50 kJ mol−1 with varying values of Ω. For example, the reactions with the two lowest ∆H‡ also show the lowest Ω. To be completely certain of the dominant reaction product, a kinetic analysis is required. Despite such an analysis being carried out in Sect. 3.1.2, a qualitative assessment of the influence of quantum tunneling can be made from the intrinsic reaction coordinate (IRC) profiles. Such IRC profiles can be visualized in Fig. 2. From them, we observe that both Reactions (R1) and (R3) have wider barriers than Reactions (R2) and (R4). Besides, both H- addition Reactions, (R2) and (R4), are also more exothermic, which also has an impact on the tunneling probability. Overall, based on the energetics and Tc (Eq. (1)) it is not possible to determine which is the dominant product of the reaction. Kinetic simulations such as the ones in Sect. 3.1.2 are necessary to discriminate them.
Reaction enthalpies (∆HR), activation energies (∆H‡), the absolute value of the imaginary frequency of vibration (Ω), and the crossover temperature (Tc) for the reactions of CH3CHO with H.
 |
Fig. 2 IRC profiles for Reactions (R1)–(R4) (We omit reaction (R5) due to the high ∆H‡). IRC profiles are presented at the rev-DSD- PBEP86(D4)/jun-cc-pV(T+d)Z not corrected by ZPVE, double-level CCSD(T), or implicit water ice environment (CPCM). |
 |
Fig. 3 Arrhenius plot of the hydrogenation rate constants (kH) for Reactions (R1)–(R4) including (dashed lines) and not including (solid lines) an implicit solvation model. All rate constants have been corrected for the implicit surface. Rate constants are presented until the lowest temperature achievable for each instanton in the sequential cooling scheme. |
3.1.2 Kinetic analysis and reactions with deuterium
We began our instanton calculations using the sequential cooling scheme at a temperature of approximately 0.7Tc, starting from a closed instanton path consisting of 36 beads. As is described in Sect. 2, we progressively converged the instanton paths at lower temperatures. To ensure convergence with respect to the number of beads at low temperatures, we increased the number of beads to 70 for temperatures below approximately 140 K in all reactions. The hydrogenation rate constants, kH, are presented in Fig. 3. We omitted the calculation of rate constants for Reaction (R5) because instanton calculations are computationally demanding, and this reaction has too high an activation energy to be competitive. The rate constants were computed down to different lower temperature limits for each reaction. This variation arises because the convergence of instanton paths depends on the reaction, with the abstraction on the CH3 moiety being particularly challenging to converge. However, in the cases we studied, the rate constants are converged or close to the asymptotic tunneling limit, so kH(Tlast) ≈ kH(10). Since the reaction outcome is dominated by Reaction (R1), the lack of rate constants at temperatures below 50 K for Reactions (R3) or (R4) does not affect our conclusions. It is worth noting that the effect of the implicit solvation scheme varies among different reactions, with Reactions (R1) and (R4) showing the most significant changes. Specifically, at low temperatures, these reactions are slower when considering an implicit water matrix due to the increase in  found for the reactions. Nevertheless, this increase does not alter the overall kinetics of the system regarding competitive reaction pathway’s.
found for the reactions. Nevertheless, this increase does not alter the overall kinetics of the system regarding competitive reaction pathway’s.
A quick inspection of Fig. 3 reveals that Reaction (R1) is the fastest across all temperatures, consistently dominating all other reaction pathways. Furthermore, aside from Reaction (R1), all other reactions exhibit rate constants that are competitive with hydrogen atom diffusion (Asgeirsson et al. 2017; Senevirathne et al. 2017), with diffusion rates (kDiff) in typical potential sites ranging from 103 to 105 s−1 at 10 K (see, for example, Fig. 7, bottom panel of Senevirathne et al. 2017). This competition suggests that even if a pre-reactive complex favoring reactions other than Reaction (R1) is formed – perhaps due to a favorable orientation of the radical attack – diffusion away from the reaction site in a reaction-diffusion competition (Chang et al. 2007) will either completely or partially suppress these alternative reaction pathways. Consequently, we conclude that Reaction (R1) is the sole outcome of the CH3CHO + H reaction. This conclusion allows us to rationalize the results obtained in our experiments (see Sect. 3.1.3).
We also examined deuteration rate constants, kD. For these calculations, we focused on the H1 and C2 positions. For example:
 (R6)
(R6)
 (R7)
(R7)
because they have the lowest ∆H‡ and the highest kH. The rate constants are shown in Fig. 4. In both cases, we observe a reduction of kD over kH due to the reduced efficiency of quantum tunneling. The kinetic isotope effects (KIEs; calculated as kH/kD) at 50 K4 that we observe according to our calculations are KIER1/R6 = 34. 7 and KIER2/R7 = 734.1, calculated including the implicit solvent approach. The KIE is larger in the case of Reaction (R7), because this reaction is a D addition and quantum tunneling is reduced, compared to Reaction (R6), which is an H abstraction and which has a higher tunneling probability.
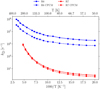 |
Fig. 4 Similar to Fig. 3 but including deuterium as an incoming particle (kD) for Reactions (R6) and (R7) |
 |
Fig. 5 Potential energy scan for Reactions (R8) (blue curve), (R9) (red curve), and (R10) (green curve) at the revDSD-PBEP86(D4)/jun-cc- pV(T+d)z level using a broken symmetry formalism. The discontinuities in r(H-H3) and r(H-O4) are caused by the insufficient resolution of the PES scan. The small bump in r(H-O4) is attributed to the constraints in the C-H bonds required to carry out the scan (see text). |
3.1.3 Hydrogenation of the CH3CO radical
The Reaction CH3CHO + H leads almost exclusively to CH3CO. Therefore, the CH3CHO reaction network must be completed by relying on the chemistry of such a radical. In this section, we study the hydrogenation of CH3CO following a scheme similar to the one shown in Fig. 1 (only this time without the H1 position). The study of the CH3O + H reaction has the peculiarity that it should be studied in the open-shell singlet channel; that is, using a biradical wavefunction. Radical-radical reactions generally show lower barriers than the radical-closed shell reactions, so we first checked which of the following reactions are barrierless using PES scans:
 (R8)
(R8)
 (R9)
(R9)
 (R10)
(R10)
 (R11)
(R11)
The scans for the barrierless reactions are shown in Fig. 5. Of all the above reactions, only Reaction (R11) was not found to be barrierless. All of the other reactions show a barrierless profile. We highlight the difficulty of obtaining clean scans for these, except for Reaction (R8), because in the latter case there are no competing channels in the direction of the scan. The other scans have discontinuities that we attribute to a combination of a low resolution and hindered conformational changes from competitive barrierless pathways during the coordinate scan. For example, the scan for Reaction (R10) shows a small bump at about 1.4 Å. This is due to the fact that in this scan we had to constrain the C-H (CH3 moiety) bond distance to prevent the optimizer from visiting the barrierless Reaction (R9) at long distances. This results in a small stiffness of the structure leading to this bump. Our calculations still predict the barrierless reaction. We recall that the calculations involving these reactions were carried out using a broken-symmetry formalism, at the revDSD-PBEP86(D4)/jun-cc-pV(T+d)z level, without correction with coupled-cluster methods. This, combined with the approximate nature of the CPCM implicit solvent scheme, makes the second set of calculations less accurate than the ones shown in Sect. 3.1.1. The energetic values for the reactions are collected in Table 2. From the table, we can see that the ∆H‡ forReaction (R11) (counterpart of (R5)) is significantly lower in this second set than in the previous reactions for the hydrogenation of acetaldehyde. We also observe the effect of the implicit solvation correction on some energetic parameters of the reaction. The abundant presence of CH4 and its isotopologs in our experiments supports the Reaction (R11), even with a barrier (see next paragraph and Sects. 3.2.1 and (4.1)). Although our results for this reaction are qualitative, they are in agreement with the experiments. However, for a more detailed study of (R11), we suggest using multireference methods. All other reactions are barrierless. Finally, it is also worth noting that our results in this section are not affected by deuteration, since tunneling does not determine the viability of the reaction (for Reactions (R8)–(R10)) or contribute significantly (Reaction (R11), which requires the fragmentation of a C-C bond).
The possibility of finding products beyond the ones obtained in barrierless channels can be easily rationalized if we consider the typical reaction-diffusion competition scheme (Chang et al. 2007). Assuming that the desorption of both reactants and the diffusion of CH3CHO are negligible, the rate constant for the reaction under this formalism is (Chang et al. 2007):
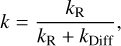 (2)
(2)
where “R” and “Diff” represent reaction and diffusion, respectively. In the anisotropic potential created by an amorphous polar environment such as ASW, the orientation from which H atoms approach is random. Assuming that all directions are equally possible for Reactions (R8)–(R11), Eq. (2) shows that if each reaction starts from different positions, as is the case, then moving out of the reaction center requires at least one diffusion step. If kR is much larger than kDiff, then the reaction can start from that orientation, even if a barrierless channel is available. This is essentially different from gas phase reactivity. A scheme for this explanation is shown in Figure 6. The arguments based on reaction-diffusion competition are therefore able to rationalize the experimental results in the light of the theoretical calculations, as we show in Sect. 4.1. However, not all reaction orientations are equally weighted, resulting in different branching ratios for reactions. Determining the exact branching ratios for these barrierless reactions from theoretical calculations is challenging. The same arguments presented in this section apply to the reactions studied in Sect. 3.1.1 (Reactions (R1)- (R5)), but in this case reactions other than Reaction (R1) are competitive with diffusion, kR ~ kDiff, and therefore Reaction (R1) is dominant.
Our theoretical results for the hydrogenation of CH3CO are in reasonable agreement with the ones presented in Ibrahim et al. (2022), although several differences appear. In their study, Ibrahim et al. (2022) indicate that the Reaction (R11) is barrierless, while the Reaction (R9) has an activation energy of ~18 kJ mol−1. This tendency contradicts our calculations and experiments. However, this discrepancy does not change the picture of both works, except for the absence of H2CCO by Reaction (R9), which we found to bevery fast, and which is supported by experiments, since all other reactions remain very fast in both our works. We believe that the reason for the discrepancy could be a non-biradical wavefunction in the case of Ibrahim et al. (2022). Starting our calculations from an ionic wavefunction, we can reproduce the results of Ibrahim et al. (2022) for the Reaction (R9) with even higher activation energies (~30 kJ mol−1).
 |
Fig. 6 Schematic view of the CH3CO + H reaction. Depending on the initial configuration of the H atoms, the different kR must compete with diffusion, kDiff. If kR ≫ kDiff, then the reaction would dominate even in the presence of a barrierless channel, as is shown in the figure. BL means barrierless. |
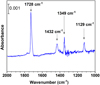 |
Fig. 7 Fourier-transform infrared profile of CH3CHO on c-ASW at 10 K. |
3.2 Experiments
3.2.1 Hydrogenation experiments
Figure 7 shows an FTIR spectrum of the solid CH3CHO on c- ASW at 10 K. The most intense absorption peak at 1728 cm−1 was attributed to the C-O stretching band (νsC=O) in CH3CHO. Additionally, other infrared bands were observed at 1432, 1349, and 1124 cm−1 can be assigned to the CH2 scissors, CH3 symmetric deformation, and C-C stretch of the solid CH3CHO, respectively (Bennett et al. 2005; Bisschop et al. 2007).
Figure 8a shows the difference spectrum of solid CH3CHO after exposure to H atoms for up to 2 hours compared to the initial spectrum of unexposed CH3CHO. The intensity of the C-O stretching band at 1728 cm−1 decreases with atom exposure time, indicating the loss of CH3CHO. Figure 8b shows variations in the relative abundance of CH3CHO after exposure to H atoms at 10 K with relevance to atom exposure times, with a blank experiment for H2 exposure. The consumption of CH3CHO was estimated to be only 10% relative to the initial amount of CH3CHO after exposure to H atoms for up to 2 hours, while no decrease was observed after exposure to H2 molecules. We expect the formed CH3CO radicals from Reaction (R1) to further react with H atoms on the surface according to the Reactions ((R8) – (R11)). Unfortunately, we could not observe any distinct infrared features for products from the pre-deposition of CH3CHO and H atoms on c-ASW at 10 K, probably because of the small amount of products. The loss of CH3CHO may be partially due to reactive desorption. We cannot quantitatively evaluate this in any of our experiments, even considering the low desorption energy of CH3CHO of ~3500 K (Molpeceres et al. 2022b; Ferrero et al. 2022), something we discuss in more detail in Sect. 4.2.
The TPD experiments demonstrate the formation of CH4 and CO based on the desorption peak observed at m/z = 16 and m/z = 28 in the temperature range of 30–60 K and 25–35 K, respectively (Fig. 9, top and middle panels). These results are in agreement with the computational predictions for the Reaction ((R11); see Sect. 3.1.3) and Ibrahim et al. (2022) experiments. In addition, we observe a desorption peak at m/z = 46 in the desorption temperature range 150–165 K (Fig. 9, bottom panel), suggesting the formation of ethanol (C2H5OH). This ethanol should be formed by the H addition reaction of CH3COH, which was obtained from the Reaction (R10), followed by:
 (R12)
(R12)
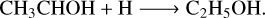 (R13)
(R13)
No distinct IR features of the products of the reaction between CH3CHO and H atoms are observed in the predeposition experiments, probably due to the small amount of solid CH3CHO (1 ML). In order to identify the products of the reaction between CH3CHO and H atoms in more detail by FTIR, we performed an additional experiment in which 30 ML of CH3CHO was co-deposited with H atoms on the Al substrate at 10 K, a technique suitable for identifying reaction products. Figure 10 shows an FTIR spectrum obtained after co-deposition of CH3CHO with H atoms on Al substrate at 10 K. In contrast to the behavior of the pre-deposited CH3CHO with H atoms (Fig. 8a), various peaks appear after the co-deposition of CH3CHO with H atoms. The appearance of a sharp peak at 2133 cm−1 was assigned to CO (Gerakines et al. 1995). After its formation on the surface, CO should be further hydrogenated to give formaldehyde (H2CO) and methanol (CH3OH), with peaks at 1724 and 1500 cm−1 for H2CO (Hidaka et al. 2004) and 1044 cm−1 for CH3OH (Nagaoka et al. 2007), as is observed in the spectrum (Fig. 10). A strong peak observed at 1305 cm−1 is attributed to CH4, which is in good agreement with the CH stretching band of CH4 (Qasim et al. 2020). In addition, two small peaks observed at 1081 and 1050 cm−1 are attributed to the CCO asymmetric bending and C–O stretching bands of CH3CH2OH, respectively (Hudson 2017).
Finally, the presence of ketene (H2CCO) is also observed in our co-deposition experiments. We could not detect it in the IR because H2CCO is easily hydrogenated (Ferrero et al. 2023), and its characteristic vibrational frequency (at 2133 cm−1) overlaps with the absorption of CO. However, clear evidence for the presence of H2CCO in the reaction mixture is observed in our TPD experiments. The desorption peaks corresponding to m/z = 42 and 14 at about 75–90 K and 150–165 K (Fig. 11) can be derived from H2CCO itself and its fragment (i.e., CH2), respectively. The detection of these m/z = 14 (CH2) and 42 (H2CCO) features in the TPD experiments, consistent with previous studies (Maity et al. 2015; Chuang et al. 2020; Fedoseev et al. 2022), provides strong evidence that H2CCO was formed under the current experimental conditions, as is supported by our theoretical models.
 |
Fig. 8 (a) Variation in the difference spectra of the solid CH3CHO after exposure to H atoms for 3, 60, and 120 min at 10 K. (b) Relative abundance of CH3CHO following a 2-hour exposure to H atoms (blue circles) compared to that with H2 molecules (red squares) on c-ASW at 10 K. |
 |
Fig. 9 TPD-QMS profile of pre-deposited CH3CHO with H atoms (black line) comparable with H2 molecules (blue line) on c-ASW at 10 K: m/z = 16 (top panel), m/z = 28 (middle panel), and m/z = 46 (bottom panel). |
 |
Fig. 10 FTIR spectrum of codeposition of CH3CHO and H atoms on Al at 10 K. |
 |
Fig. 11 TPD-QMS profile for the m/z = 14 and 42. Black arrows correspond to features attributable to H2CCO, obtained after the reaction of the pre-deposited CH3CHO with H atoms on c-ASW at 10K. The desorption peaks at 100–130 K and 140–150 K are derived from the remaining CH3CHO on c-ASW after exposure to H atoms. The peaks at 75–90 K and 150–165 K represent the desorption of H2CCO (Maity et al. 2015; Chuang et al. 2020; Fedoseev et al. 2022). |
3.2.2 Isotopic labeling
Figure 12 shows the evolution of the difference spectra of CH3 CHO after exposure to D atoms for up to 2 hours as well as that of CH3 CHO without atomic exposure. Similar to the behavior after exposure to H atoms, the C-O stretching band of CH3CHO at 1728 cm−1 decreases with exposure times, indicating the loss of CH3CHO after interaction with D atoms on the Al substrate. Unfortunately, we cannot identify clear FTIR features for the products of the pre-deposited CH3CHO with D atoms.
On the other hand, we observe various features for the formation of isotopologs of the products based on the TPD experiments. Figure 13 shows the TPD spectrum after the reaction of CH3CHO with D atoms on A1 substrate at 10 K. Since CH3CHO desorbs at the temperature between 90 and 145 K and shows a CHO fragment (m/z = 29) upon ionization, the desorption peak for m/z = 30 (the CDO fragment) is attributed to the deuterated counterpart of acetaldehyde, CH3CDO. Thus, the desorption peak (m/z = 30 – top panel) observed at the same temperature after exposure to D atoms identifies the formation of CH3CDO. The formation of CH3CDO provides compelling evidence for the reaction between CH3CO and D atoms: CH3CO +D → CH3CDO. This process, in which the CH3CO radical is produced by the Reaction (R6), is consistent with the calculated results.
Furthermore, the desorption peaks at m/z = 17 and 18 (Fig. 13, middle and bottom panels) within the temperature range of 35–65 K are indicative of the formation of deuterated methane isotopologs, CH3D and CH2D2. These species could be produced by the reactions in (R16), as discussed in Sect. 4.1, although we cannot be certain due to the high barriers associated with H abstraction in CH3D (Lamberts et al. 2022).
A tiny peak corresponding to deuterated methanol (m/z = 36; CD3OD) is observed to desorb at about 135 K (Fig. 14). The formation of CD3 OD has been proposed to result from the deuteration of CO (Watanabe & Kouchi 2002), implying that the CO molecule is likely produced by the reaction of CH3 CO (Reaction (R11)) with H or D atoms under the current experimental conditions, a finding consistent with our calculated results. In addition, we also observe the desorption peaks at m/z = 49 and 50 in the temperature range of about 130–200 K. These peaks can be attributed to the formation of ethanol isotopologs corresponding to CH3 CD2OD and/or CH2DCD2OD (see Figure 15), which were obtained by reactions of CH3CO radicals with D atoms, followed by:
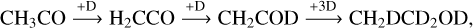 (R14)
(R14)
and
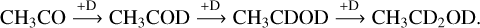 (R15)
(R15)
The formation of ethanol isotopologs is consistent with the computational results obtained from Reactions (R9) and (R10) (see Sect. 3.1.3).
The appearance of a desorption peak observed at m/z = 48 (see Fig. 16) likely indicates the formation of CH3CHDOD, resulting from the addition reaction of D to CH3CHO under the current experimental conditions. Nevertheless, based on the quantitative analysis of desorption areas for the reaction products, CH3CHDOD is found to be a minor species in the reaction between CH3CHO and D atoms. The predominant reaction involves CH3CO radicals with D atoms, leading a higher yield of different products.
 |
Fig. 12 Variation in the difference spectra of the solid CH3 CHO on the Al substrate at 10 K after exposure to D atoms for 10, 60, and 120 min comparable with the initial CH3CHO. |
 |
Fig. 13 TPD-QMS profile of pre-deposition of CH3 CHO with D atoms (black line) comparable with D2 (blue line) on Al substrate at 10 K, m/z = 30 (top panel); m/z = 17 (middle panel), and m/z =18 (bottom panel). The m/z = 30 (top panel) in the blue line represents the fragmentation of CH3CHO. |
 |
Fig. 14 TPD-QMS profile of m/z = 36 observed for the pre-deposition of CH3 CHO and D atoms (black line) is compared with that of D2 molecules (blue line) on the A1 substrate. |
 |
Fig. 15 TPD-QMS profiles of m/z = 49 and 50 (black lines) desorbed at around 130–200 K, likely corresponding to ethanol isotopologs CH3CD2OD and/or CH2DCD2OD. These species were yielded through the reaction of CH3CHO and D atoms, with behavior comparable to that of D2 molecules (blue line) on Al substrate at 10 K. |
 |
Fig. 16 TPD-QMS profile for the desorption peak observed at m/z = 48 (black line) may be attributed to the formation of CH3CHDOD through the addition reaction of D to CH3CHO. This is in the comparison to the desorption behavior for D2 (blue line) on the Al substrate at 10 K. |
4 Discussion
4.1 Merging theory and experiments: Comparison with previous works
Once the theoretical and experimental results have been discussed, we are in a privileged position to discuss the chemical mechanism behind the surface hydrogenation of acetaldehyde. The main experimental finding comes after the analysis of the 1432, 1349, and 1124 cm−1 IR decay bands that, after comparison with the amount of deposited CH3CHO, reveals a limited destruction of around of a 10%, with the rest of the CH3CHO either unaffected or reformed after a H-abstraction and H- addition cycle. This finding is the easiest to rationalize based on the quantum chemical calculations. The limited destruction or conversion of CH3CHO is due to the dominance of the (R1) channel in the CH3CHO + H/D reaction. Previous literature studies on the hydrogenation of aldehydes (Mondal et al. 2021) have not considered the H-abstraction reactions, perhaps analogously to the limited importance of this type of reaction in the parent molecule formaldehyde, H2CO (Song & Kästner 2017). However, for CH3CHO we found otherwise, with H-abstraction dominating the reactivity. Our deuteration experiments show a tiny fraction of CH3CHDOD, indicating the possibility also of some hydrogen or deuterium addition in the hydrogenation or deuteration of CH3CHO. This is somewhat coherent with the rate constants for H addition found in Reactions (R2) and (R7), when CH3CHO and H are in deep binding sites with slow diffusion rates (see Fig. 6 and associated discussion). However, H addition is a minor channel of the reaction, whose quantification to the total branching ratio is difficult. Therefore, in Appendix B we recommend considering a 1.0 branching ratio toward H abstraction (Reaction (R1)).
After Reaction (R1), any subsequent hydrogenation on the new system involves the very reactive CH3CO radical. From a theoretical standpoint, we determined that all reaction positions are, in principle, possible. Beginning with Reaction (R8), the H (or D) addition on this position reforms CH3CHO and, based on the experiment, is the most likely outcome, with 90% of the reactive events in this direction, determined from the total fraction of CH3CHO remaining after concluding our experiments. However, neither our calculations nor our experiments can rule out the contribution of the reforming of CH3CHO from secondary reactions; that is, hydrogenation of H2CCO (see below and Ibrahim et al. 2022; Ferrero et al. 2023; Ibrahim et al. 2024). For Reactions (R9), (R10), and (R11), the products are H2CCO (and H2), CH3 COH (methyl-methoxy carbene; Schreiner et al. 2011), and CH4 + CO. Beginning with H2CCO, as we mention in Sect. 3.2, we cannot identify it in the IR spectrum because its main transition falls at 2133 cm−1, overlapping with the CO band. However, the appearance of additional desorption features for m/z = 14 (CH2) and 42 (H2CCO) between 75–90 and 150– 165 K in the TPD experiments (Fig. 11) is in line with previous studies (Maity et al. 2015; Chuang et al. 2020; Fedoseev et al. 2022), which confirmed the desorption of H2CCO. Nevertheless, Ferrero et al. (2023), using instanton calculations such as the ones shown in this work, show that the H2CCO hydrogenation is very efficient with final products CH3CHO and perhaps C2H5OH (ethanol). Bisschop et al. (2007); Ibrahim et al. (2022) also report CO + CH4 and H2CO as products of the H2CCO + H reaction, which is also coherent with our results. Combining experiments and theory, we determined that H2CCO must be a product of the CH3CHO hydrogenation under interstellar conditions, but in small amounts. Continuing with CH3COH, even though it is formed in our calculations, we are unable to comment much on this species from an experimental point of view. This is due to the limited experimental data, but also to its transient nature. As a reactive carbene, we expect that the CH3COH+ 2H→ C2H5OH reaction will dominate in our experimental setup. Indeed, C2H5OH is seen in our co-deposition experiments, and the experimental and theoretical synergy carried out in this work allowed us to conclude that it is a secondary product of the hydrogenation of CH3CHO rather than a direct one. The presence of C2 H5 OH is puzzling when checking the previous literature. In the experiments of Fedoseev et al. (2022) (for H2CCO + H) C2H5OH is seen, as in our experiments. However, Ibrahim et al. (2022) and Ibrahim et al. (2024) do not report its presence after the same reaction. The reason for this disagreement is likely due to the experimental conditions, and our results provide a reconciling scenario. We could not detect C2H5OH using the IR spectra in our pre-deposition experiment, which would be the one directly comparable to the experiments of Ibrahim et al. (2022) needing to rely on TPD-QMS measurements (see Fig. 9) for that. However, the presence of C2H5OH is seen in our co-deposition experiments starting from CH3 CHO mimicking Fedoseev et al. (2022). Although other experimental conditions differ between our setups, such as the H flux or irradiation time, we think the reason for the disagreement stems from the deposition strategy.
Finally, the last experimental product that we observe is CH4 (and its isotopologs, see below) and CO. Given the enormous ∆H‡ found for Reaction (R5), the formation of CH4 must come from the hydrogenation of CH3CO, or perhaps of further radicals in the hydrogenation sequence of CH3CO, out of the scope of the present paper. However, we find it likely that CH4 comes directly from CH3CO via Reaction (R11). CO is seen in our experiments, either directly as CO or as its hydrogenated products H2CO and CH3OH as discussed above.
Our deuteration experiments do show a variety of isotopologs of the products discussed in the previous paragraphs. The presence or absence of certain isotopologs not only aids our experiments but also allows us to confirm our theoretical observations. For CH3 CHO + D, our experiments reveal the majoritarian formation of CH3CO that later reforms CH3CDO. Likewise, we clearly see the formation of CH3D and CH2D2, which might arise from the sequence of reactions (omitting HD when applicable):
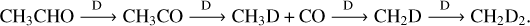 (R16)
(R16)
These reactions serve as a proxy to suggest that the CH3D + H/D→ CH2D + H2 / HD could be a viable reaction, although we cannot confidently ensure it, because some reaction steps in (R16) have high barriers. A preliminary search at the rev-DSD- PBEP86(D4)/jun-cc-pV(T+d)Z level for the CH3D + D → CH2D + HD (The third reaction in the scheme of reactions shown in (R16)) shows that the reaction is slightly exothermic, unlike the full hydrogenated counterpart, by ∆HR = −3.3 kJ mol−1 (−404 K), making the reaction scheme (R16) in principle viable albeit probably very slow. Overall, while the presence of CH3D is easy to explain from experiments and theory, the presence of CH2D2 is not that easily explained, with secondary and tertiary hydrogenation branches being important, and requiring a careful validation out of the scope of this work. The CO formed in the previous reaction can also be hydrogenated to fully deuterated methanol (CD3OD) (Watanabe & Kouchi 2002), a reaction that could not happen without the formation of CO, further increasing our confidence in the viability of Reaction (R11). We also observe clear signals of the formation of ethanol isotopologs, especially of CH3CD2OD or CH2DCD2OD (Reaction scheme in (R14) and (R15) and Fig. 15), once again merging experimental and theoretical results and highlighting the importance of Reaction (R1) as the initiator of the complex chemistry observed in the experiments. We reiterate that a small fraction of CH3CHDOD is also found (Fig. 16).
We briefly comment on the differences between our new set of experiments and the classical ones of Bisschop et al. (2007) that inspired this work. In both our experiments, we obtain the same products of the reaction, although in different proportions. While we only find a conversion of CH3CHO equal to 10% for the duration of our experiment, Bisschop et al. (2007) report variable conversion rates, varying from between 30% to almost total conversion (see their Table 5). These quantitative differences should come from different factors, the most likely reasons being the temperature of the H atoms and the selection of the substrate. In our setup, the H atoms are cooled down before hitting the surface, guaranteeing immediate thermalization on the surface. In Bisschop et al. (2007), the H-atoms were introduced in the chamber at 300 K, which can promote a plethora of reactions through different reaction mechanisms, such as the Eley-Rideal or Harris-Kasemo ones. Moreover, in our setup, we carried out our experiments on ASW, where the interactions between CH3CHO molecules with the surface will be different than in pure CH3CHO or than in a metallic substrate.
Lastly, in light of what has been presented in this section, we have prepared a series of recommendations to include this reaction in astrochemical rate equation models. We have gathered our conclusions in Appendix B.
4.2 Astrophysical implications
Our results carry a series of implications for the astrochemical community, in particular regarding the deuteration and reactive desorption of CH3CHO. Acetaldehyde is a molecule routinely detected in a variety of interstellar environments (Cazaux et al. 2003; Occhiogrosso et al. 2014; Walsh et al. 2014; Imai et al. 2016; Codella et al. 2015; Holdship et al. 2019; Bonfand et al. 2019), including several detections in prestellar cores (Bacmann et al. 2012; Cernicharo et al. 2012; Scibelli et al. 2021; Jiménez-Serra et al. 2016; Megías et al. 2022), where there is little energy for bringing the molecules to the gas phase, where they are detected. The formation of CH3CHO is debated, including the gas-phase route C2H5 + O → CH3CHO + H (Vazart et al. 2020) or ice surface routes. These routes involve the radical-radical recombination of HCO + CH3, on ice (Garrod 2013; Molpeceres et al. 2024a; Lamberts et al. 2019), although on water ice this route is sometimes considered less effective (Enrique-Romero et al. 2021). Alternative routes include the radiolytic (Shingledecker et al. 2018) or nonthermal formation (Jin & Garrod 2020). Regardless of the formation route, CH3CHO will form or deplete on the grain, and it is at this stage when the title reaction can take place.
Our results show that acetaldehyde is mostly processed and reformed through Reactions (R1) and (R8), and, although other products and routes exist, as is extensively discussed in this manuscript, the astrophysical implications of our work are mainly carried by this limited destruction. We shall focus on those in this section. First, we shall talk about reactive desorption. When an H atom is abstracted from the HCO moiety in CH3CHO, the hydrogenation of the resulting CH3CO radical could trigger a chemical desorption event, considering that the binding energy of CH3CHO is among the lowest for COMs. As we indicated in Sect. 3.2, we cannot confirm this possibility, as the different hydrogenation branches opened from our reaction make it very difficult to confirm or deny the possibility of chemical desorption. Optimistic estimates of the probability of chemical desorption for COMs place the probability per reaction event at around 1% (Garrod et al. 2007). Because this amount should be included in the 10% of nonreformed CH3 CHO, it is experimentally extremely hard to prove. Yet, CH3 CHO constitutes the best-case scenario with an effective H-abstraction/H-addition cycle, the high reaction energy for the CH3 CO + H reaction, and the relatively low binding energy on water ice (Molpeceres et al. 2022b; Ferrero et al. 2022).
Another important implication of our work pertains to the formation of ethanol, C2H5OH, which is predicted to be formed in very low amounts from Reaction (R2), but actually from the evolution of the CH3 CO + H reaction. This is in contrast with the hydrogenation of formaldehyde (H2CO), the prototypical interstellar aldehyde, which leads to methanol CH3OH (Watanabe & Kouchi 2002; Hidaka et al. 2004). In Mondal et al. (2021), the authors indicate that the connection between CH3CHO and C2H5OH through direct hydrogenation is not enough to account for the abundances of C2H5OH in the hot core G10.47+0.03, and that their models reveal that CH3 + CH2OH → C2H5OH is needed to explain the observations. Our results fully adhere to this picture and strongly suggest that the link between CH3CHO and C2H5OH is not straightforward.
Finally, we reserve some words for the possibility of finding deuterated acetaldehyde. A relatively recent publication Jørgensen et al. (2018) shows that CH3CDO is the isotopolog of a COM with the highest ratio to the non-deuterated molecule, with a percentage of 8% in IRAS16293–2422. This result reinforces the results obtained in this work and suggests as a possible explanation for this observation the H-abstraction or H-addition route discussed in this work, in addition to the explicit detection of CH3CDO in our experiments. Nevertheless, it is important to remark that other CH3CHO isotopologs, CH2DCHO and CD2HCHO, have also been detected in the same source (Coudert et al. 2019; Ferrer Asensio et al. 2023). These last findings do not rule out the importance of our reaction routes but show the complexity associated with deuterium fractionation in the ISM, where gas and surface chemistry are in constant interplay.
5 Conclusions
We conclude by providing a series of bullet points that summarize our work.
Our calculations and experiments show that the CH3CHO + H does not lead to significant conversion. The reason for that limited conversion is found in the dominance of hydrogen abstraction in the HCO moiety of CH3CHO, which in turn leads to facile reforming of CH3CHO. Quantifying this conversion leads us to a maximum of 10% of CH3CHO under our experimental conditions.
Other minor products of the reaction of CH3CHO with H come from the hydrogenation of the CH3 CO reactive radical and are H2CO, CH3OH, C2H5OH, H2CCO, CO, or CH4, revealing a very complex chemistry. In general, our calculations and previous knowledge of the chemistry of the different intermediates found in our calculations can satisfactorily explain the different products that we obtain. However, and because the study of the CH3 CO + H is more qualitative due to theoretical constraints, more sophisticated approaches are required to derive accurate branching ratios for the reaction.
Our experiments reveal the effective formation of CH3CDO, supporting the idea of a H/D-abstraction and H/D-addition cycle as the major route for the CH3CHO + H reaction.
Although we can explain the vast majority of our experimental observations with our calculations, the presence of doubly deuterated methane CH2 D2 and the CH3CHDOD isotopolog in our deuteration experiments remains difficult to explain. In general, several hydrogenation and deuteration branches of the minor products could lead to different products, making explaining the complete reaction network a very difficult task.
Our results align with the results found in the modeling literature (Mondal et al. 2021) suggesting that the link between acetaldehyde and ethanol is not direct from the former to the latter via hydrogenation.
We are unable to determine whether reactive desorption happens for CH3CHO, although in light of the energetic parameters found by our theoretical calculations, summed to the H-abstraction and addition cycle present in the reaction, we consider it a likely outcome.
Our experiments and calculations nicely explain the excess of CH3CDO found in the IRAS 16293-2422 hot core (Jørgensen et al. 2018). However, we remark that a single chemical reaction is not enough to explain complex deuterium enrichment processes in space.
Future avenues of the present work could address the presence of certain non-easy-to-explain isotopologs in our experiments, or the study of the impact of the quantities derived here in realistic astrochemical models. Likewise, we are currently tackling the complex problem of CH3 CHO chemical desorption using more sophisticated molecular dynamics approaches. We shall continue combining experiments and calculations to fully unravel complex interstellar reaction networks.
Data availability
The Cartesian coordinates for the structures shown in this paper, as well as the numerical values for the rate constants, can be retrieved at https://zenodo.org/records/14278652. Any further data can be requested from the corresponding authors.
Acknowledgements
The authors thank the anonymous reviewer for the thorough revision of the manuscript. We thank Professor Johannes Kästner at Stuttgart University for providing a development version of CHEMSHELL. We also thank Dr. Masashi Tsuge and Dr. Hiroshi Hidaka for fruitful discussions on the experimental results. G.M acknowledges the support of the grant RYC2022-035442-I funded by MICIU/AEI/10.13039/501100011033 and ESF+. G.M. also received support from project 20245AT016 (Proyectos Intramurales CSIC). We acknowledge the computational resources provided by bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster), the DRAGO computer cluster managed by SGAI-CSIC, and the Galician Supercomputing Center (CESGA). The supercomputer FinisTerrae III and its permanent data storage system have been funded by the Spanish Ministry of Science and Innovation, the Galician Government and the European Regional Development Fund (ERDF). Y.O and N.W. acknowledge the funding support from JSPS KAKENHI grant nos. JP23H03980, JP21H04501, and JP22H00159.
Appendix A Determination of reaction barriers using a small 2 H2O cluster model. Validity of the implicit surface approximation
In the main text, we obtain the reaction descriptors and subsequent kinetic rate constants using two implicit approximations. In addition to an implicit solvation approach, we assume that the ASW surface does not play a major role in the reaction, introducing surface effects via rotational partition function fixing. These approximations are required for including nuclear quantum effects in the calculation of our rate constant while keeping an accurate potential (CCSD(T)/aug-cc-pVTZ//rev- DSD-PBEP86(D4)/jun-cc-pv(T+d)Z). The applicability of these approximations is ensured by a single condition, that the reaction descriptors are not affected by the water matrix. Such a condition is fulfilled normally in adsorbates bound to ASW via weak physisorption forces, although it has been applied to molecules as tightly bound as OH (Meisner et al. 2017; Lamberts & Kästner 2017a,b; Molpeceres & Kästner 2021; Molpeceres et al. 2022a; Molpeceres & Rivilla 2022; Ferrero et al. 2023). Acetaldehyde has one of the lowest binding energies found for any COM (Ferrero et al. 2022; Molpeceres et al. 2022b) so we do not expect a different behavior to the cases cited above. Nevertheless, we explicitly tested whether the introduction of explicit water affects the reaction descriptors and reaction profiles in our studied reactions. The tests we performed include comparing the activation energies, with and without water molecules,  , and
, and 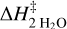 . For the 2 H2O coupled cluster calculations, we used the domain local pair natural orbital (DLPNO) (Guo et al. 2018) version of CCSD(T), using a TightPNO localization scheme.
. For the 2 H2O coupled cluster calculations, we used the domain local pair natural orbital (DLPNO) (Guo et al. 2018) version of CCSD(T), using a TightPNO localization scheme.
The results of the tests are shown in Fig. A.1 and Table A.1. Starting with the IRC profiles shown in Fig. A.1 it is evident that in all cases the barrier shapes are almost identical, with subtle differences. These are, for example, that in the 2 H2O model the IRC path extends beyond the gas phase one, a consequence of the interaction of the reacting molecules with the 2 H2O. The most important deviation appears for Reaction (R4) that, in the 2 H2O model, experiences a restructuring of the water dimer to better accommodate the CH2 CHO radical. Nevertheless, the change in the profile is still very small. Similarly, the deviations in ∆H‡ are minimal and cannot be ruled out if the deviations are a consequence of the 2 H2O model or by the DLPNO scheme used in the computation of the molecular energies. Overall, we find that including explicit water molecules does not affect our results. While the inclusion of further water molecules can also have an impact, this impact will not be larger than the one exerted by the closest water molecules. We remember that polar solvent effects are introduced in the main text utilizing an implicit solvation approach. Overall, we confidently conclude that the approximations in this work are justified in light of these results.
Appendix B Modeling recommendations
The picture left by the combination of our calculations and experiments is a complex one, despite the relative simplicity of the CH3CHO molecule. We think this is a trait common to all COMs (with more than one carbon atom) hydrogenations. As chemical complexity increases, the possibility of reaction branches starts to emerge, and hydrogenation experiments are harder to reconcile with theory because the former are suited to detect final products whereas the latter are more suited to investigate elementary processes. We feel that approaches like ours, merging experiments and theory are fundamental to securely identify representative interstellar reaction routes.
However, the here studied reactions (or reaction schemes) seek to provide reliable data to feed astrochemical models, and in the particular case of this study, COMs and deuterium fractionation reaction networks, aiming to a better description of interstellar environments. Once the complexity of a reaction scheme starts to increase, as the one shown in this work, several assumptions and recommendations for astrochemical models need to be given. In this section we provide our guidelines for the treatment of CH3CHO hydrogenation, justifying our decisions.5 In the first place, and although the most exact approach is to include the exact rate constants for (R2)-(R4) and an accurate modeling of H atom diffusion, we consider safe deactivating the reactions in reaction networks. We base our recommendation on the limited conversion that we observe for CH3CHO, which remains mostly unaltered in our experiments, an observation supported by the CH3CHO ⇋ CH3CO + H cycle that we derive theoretically. The minor presence of CH3CHDOD in our experiments, the only product that unambiguously can come from Reaction (R7) (as a deuterated proxy of Reaction (R2)) also encourages us to consider the branching ratio of reactions other than Reaction (R1) very low, and in the absence of further evidence, negligible. The second interpretation of our results concerns the branching ratios (α values) of Reactions (R9)- (R11). Rationalizing this choice is easy. If we consider that Reaction (R8) is the only reaction from the CH3CHO + H then, >the 90% of remaining CH3CHO in our hydrogenation experiment (Sect. 3.2.1) must come from Reaction (R8) alone. The remaining 10% should be distributed along the different reaction channels (R9)-(R11). It is extremely difficult to determine α for these reactions experimentally or theoretically. We consider that the best compromise is to equally weigh the remaining 10% along the three different reaction channels. Therefore we recommend setting α=0.90 for (R8) and 0.03 for (R9)-(R11).
We would like to indicate that a certain degree of chemical intuition and data interpretation is needed to provide these modeling recommendations. Because our calculations show a significant resistance of CH3CHO to hydrogenation and deuteration, we interpret our data based on this observation and the evidence from the quantum chemical calculations. Should minor channels along the hydrogenation sequence, for example, H2CCO hydrogenation reforming CH3CHO as reported in the literature (Ferrero et al. 2023; Fedoseev et al. 2022) have a larger contribution, several of the α values mentioned above can vary. Nevertheless, we expect these changes to be small.
References
- Alvarez-Barcia, S., Russ, P., Kästner, J., & Lamberts, T. 2018, MNRAS, 479, 2007 [CrossRef] [Google Scholar]
- Asgeirsson, V., Jónsson, H., & Wikfeldt, K. T. 2017, JPCC, 121, 1648 [Google Scholar]
- Bacmann, A., & Faure, A. 2014, in SF2A-2014: Proceedings of the Annual meeting of the French Society of Astronomy and Astrophysics, eds. J. Ballet, F. Martins, F. Bournaud, R. Monier, & C. Reylé, 3 [Google Scholar]
- Bacmann, A., Taquet, V., Faure, A., Kahane, C., & Ceccarelli, C. 2012, A&A, 541, L12 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Barone, V., & Cossi, M. 1998, JPCA, 102, 1995 [NASA ADS] [CrossRef] [Google Scholar]
- Bartlett, R. J., & Purvis, G. D. 1978, Int. J. Quantum Chem., 14, 561 [CrossRef] [Google Scholar]
- Bennett, C. J., Jamieson, C. S., Osamura, Y., & Kaiser, R. I. 2005, ApJ, 624, 1097 [Google Scholar]
- Bisschop, S. E., Fuchs, G. W., van Dishoeck, E. F., & Linnartz, H. 2007, A&A, 474, 1061 [CrossRef] [EDP Sciences] [Google Scholar]
- Bohlin, R. C., Savage, B. D., & Drake, J. F. 1978, ApJ, 224, 132 [Google Scholar]
- Bonfand, M., Belloche, A., Garrod, R. T., et al. 2019, A&A, 628, A27 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Caldeweyher, E., Ehlert, S., Hansen, A., et al. 2019, JCP, 150, 154122 [Google Scholar]
- Cazaux, S., Tielens, A. G. G. M., Ceccarelli, C., et al. 2003, ApJ, 593, L51 [CrossRef] [Google Scholar]
- Ceccarelli, C., Caselli, P., Bockelée-Morvan, D., et al. 2014, in Protostars and Planets VI, eds. H. Beuther, R. S. Klessen, C. P. Dullemond, & T. Henning (Tucson: University of Arizona Press), 859 [Google Scholar]
- Cernicharo, J., Marcelino, N., Roueff, E., et al. 2012, ApJ, 759, L43 [NASA ADS] [CrossRef] [Google Scholar]
- Chang, Q., Cuppen, H. M., & Herbst, E. 2007, A&A, 469, 973 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Chuang, K. J., Fedoseev, G., Ioppolo, S., van Dishoeck, E. F., & Linnartz, H. 2020, MNRAS, 455, 1702 [Google Scholar]
- Codella, C., Fontani, F., Ceccarelli, C., et al. 2015, MNRAS, 449, L11 [CrossRef] [Google Scholar]
- Coudert, L. H., Margulès, L., Vastel, C., et al. 2019, A&A, 624, A70 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Dunning, T. H. 1989, JCP, 90, 1007 [Google Scholar]
- Eckart, C. 1930, Phys. Rev., 35, 1303 [Google Scholar]
- Enrique-Romero, J., Ceccarelli, C., Rimola, A., et al. 2021, A&A, 655, A9 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Enrique-Romero, J., Rimola, A., Ceccarelli, C., et al. 2022, ApJS, 259, 39 [NASA ADS] [CrossRef] [Google Scholar]
- Fedoseev, G., Qasim, D., Chuang, K.-J., et al. 2022, ApJ, 924, 110 [NASA ADS] [CrossRef] [Google Scholar]
- Ferrer Asensio, J., Spezzano, S., Coudert, L. H., et al. 2023, A&A, 670, A177 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Ferrero, S., Grieco, F., Ibrahim Mohamed, A.-S., et al. 2022, MNRAS, 516, 2586 [CrossRef] [Google Scholar]
- Ferrero, S., Ceccarelli, C., Ugliengo, P., Sodupe, M., & Rimola, A. 2023, ApJ, 951, 150 [CrossRef] [Google Scholar]
- Fuchs, G. W., Cuppen, H. M., Ioppolo, S., et al. 2009, A&A, 505, 629 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Garcia-Ratés, M., & Neese, F. 2020, J. Comp. Chem., 41, 922 [Google Scholar]
- Garrod, R. T. 2013, ApJ, 765, 60 [Google Scholar]
- Garrod, R. T., Wakelam, V., & Herbst, E. 2007, A&A, 467, 1103 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Garrod, R. T., Jin, M., Matis, K. A., et al. 2022, ApJS, 259, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Gerakines, P. A., Schutte, W. A., Greenberg, J. M., & van Dishoeck, E. F. 1995, A&A, 296, 810 [NASA ADS] [Google Scholar]
- Gillan, M. J. 1987, J. Phys. C, 20, 3621 [NASA ADS] [CrossRef] [Google Scholar]
- Guo, Y., Riplinger, C., Becker, U., et al. 2018, JCP, 148, 011101 [NASA ADS] [Google Scholar]
- Herbst, E., & Van Dishoeck, E. F. 2009, Ann. Rev. Astron. Astrophys., 47, 427 [Google Scholar]
- Hidaka, H., Watanabe, N., Shiraki, T., Nagaoka, A., & Kouchi, A. 2004, ApJ, 614, 1124 [Google Scholar]
- Holdship, J., Viti, S., Codella, C., et al. 2019, ApJ, 880, 138 [NASA ADS] [CrossRef] [Google Scholar]
- Hudson, R. L. 2017, Spectrochim Acta A Mol Biomol Spectrosc, 187, 82 [CrossRef] [Google Scholar]
- Ibrahim, M., Guillemin, J.-C., Chaquin, P., Markovits, A., & Krim, L. 2022, PCCP, 24, 23245 [CrossRef] [Google Scholar]
- Ibrahim, M., Guillemin, J.-C., Chaquin, P., Markovits, A., & Krim, L. 2024, PCCP, 26, 4200 [NASA ADS] [CrossRef] [Google Scholar]
- Imai, M., Sakai, N., Oya, Y., et al. 2016, ApJ, 830, L37 [CrossRef] [Google Scholar]
- Jiménez-Serra, I., Vasyunin, A. I., Caselli, P., et al. 2016, ApJ, 830, L6 [Google Scholar]
- Jiménez-Serra, I., Vasyunin, A. I., Spezzano, S., et al. 2021, ApJ, 917, 44 [CrossRef] [Google Scholar]
- Jin, M., & Garrod, R. T. 2020, ApJS, 249, 26 [Google Scholar]
- Jørgensen, J. K., Müller, H. S. P., Calcutt, H., et al. 2018, A&A, 620, A170 [Google Scholar]
- Kästner, J. 2014, Wiley Interdiscip. Rev. Comput. Mol. Sci., 4, 158 [CrossRef] [Google Scholar]
- Kästner, J., Carr, J. M., Keal, T. W., et al. 2009, JPCA, 113, 11856 [CrossRef] [Google Scholar]
- Knowles, P. J., Hampel, C., & Werner, H.-J. 1993, JCP, 99, 5219 [Google Scholar]
- Kozuch, S., & Martin, J. M. 2011, PCCP, 13, 20104 [CrossRef] [Google Scholar]
- Lamberts, T. 2018, A&A, 615, L2 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Lamberts, T., & Kästner, J. 2017a, ApJ, 846, 43 [Google Scholar]
- Lamberts, T., & Kästner, J. 2017b, JPCA, 121, 9736 [NASA ADS] [CrossRef] [Google Scholar]
- Lamberts, T., Markmeyer, M. N., Kolb, F. J., & Kästner, J. 2019, ACS Earth. Sp. Chem., 3, 958 [CrossRef] [Google Scholar]
- Lamberts, T., Fedoseev, G., Van Hemert, M. C., et al. 2022, ApJ, 928, 48 [NASA ADS] [CrossRef] [Google Scholar]
- Maity, S., Kaiser, R. I., & Jones, B. M. 2015, PCCP, 17, 3081 [NASA ADS] [CrossRef] [Google Scholar]
- Megías, A., Jiménez-Serra, I., Martín-Pintado, J., et al. 2022, MNRAS, 519, 1601 [CrossRef] [Google Scholar]
- Meisner, J., Lamberts, T., & Kästner, J. 2017, ACS Earth. Sp. Chem., 1, 399 [CrossRef] [Google Scholar]
- Metz, S., Kästner, J., Sokol, A. A., Keal, T. W., & Sherwood, P. 2014, Wiley Interdiscip. Rev. Comput. Mol. Sci., 4, 101 [Google Scholar]
- Miksch, A. M., Riffelt, A., Oliveira, R., Kästner, J., & Molpeceres, G. 2021, MNRAS, 505, 3157 [NASA ADS] [CrossRef] [Google Scholar]
- Molpeceres, G., & Kästner, J. 2021, ApJ, 910, 55 [NASA ADS] [CrossRef] [Google Scholar]
- Molpeceres, G., & Rivilla, V. M. 2022, A&A, 665, A27 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Molpeceres, G., Jimenez-Serra, I., Oba, Y., et al. 2022a, A&A, 663, A41 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Molpeceres, G., Kästner, J., Herrero, V. J., Peláez, R. J., & Maté, B. 2022b, A&A, 664, A169 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Molpeceres, G., Rivilla, V. M., Furuya, K., et al. 2023, MNRAS, 521, 6061 [NASA ADS] [CrossRef] [Google Scholar]
- Molpeceres, G., Furuya, K., & Aikawa, Y. 2024a, A&A, 688, A150 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Molpeceres, G., Tsuge, M., Furuya, K., et al. 2024b, J. Phys. Chem. A, 128, 3874 [NASA ADS] [CrossRef] [Google Scholar]
- Mondal, S. K., Gorai, P., Sil, M., et al. 2021, ApJ, 922, 194 [NASA ADS] [CrossRef] [Google Scholar]
- Nagaoka, A., Watanabe, N., & Kouchi, A. 2007, JPCA, 111, 3016 [NASA ADS] [CrossRef] [Google Scholar]
- Neese, F., Wennmohs, F., Becker, U., & Riplinger, C. 2020, JCP, 152, 224108 [Google Scholar]
- Nguyen, T., Fourré, I., Favre, C., et al. 2019, A&A, 628, A15 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Nguyen, T., Oba, Y., Shimonishi, T., Kouchi, A., & Watanabe, N. 2020, ApJ, 898, L52 [NASA ADS] [CrossRef] [Google Scholar]
- Nguyen, T., Oba, Y., Sameera, W. M. C., Kouchi, A., & Watanabe, N. 2021a, ApJ, 918, 73 [NASA ADS] [CrossRef] [Google Scholar]
- Nguyen, T., Oba, Y., Sameera, W. M. C., Kouchi, A., & Watanabe, N. 2021b, ApJ, 922, 146 [NASA ADS] [CrossRef] [Google Scholar]
- Nguyen, T., Oba, Y., Sameera, W. M. C., et al. 2023, ApJ, 944, 219 [NASA ADS] [CrossRef] [Google Scholar]
- Oba, Y., Osaka, K., Watanabe, N., Chigai, T., & Kouchi, A. 2014, Fa. Di., 168, 185 [NASA ADS] [CrossRef] [Google Scholar]
- Oba, Y., Tomaru, T., Lamberts, T., Kouchi, A., & Watanabe, N. 2018, Nat. Astron., 2, 228 [NASA ADS] [CrossRef] [Google Scholar]
- Occhiogrosso, A., Vasyunin, A., Herbst, E., et al. 2014, A&A, 564, A123 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Papajak, E., Zheng, J., Xu, X., Leverentz, H. R., & Truhlar, D. G. 2011, J. Chem. Theory Comp., 7, 3027 [Google Scholar]
- Perrero, J., Ugliengo, P., Ceccarelli, C., & Rimola, A. 2023, MNRAS, 525, 2654 [NASA ADS] [CrossRef] [Google Scholar]
- Purvis, G. D., & Bartlett, R. J. 1982, JCP, 76, 1910 [Google Scholar]
- Qasim, D., Fedoseev, G., Chuang, K. J., et al. 2020, Nat. Astron., 4, 781 [Google Scholar]
- Rimola, A., Taquet, V., Ugliengo, P., Balucani, N., & Ceccarelli, C. 2014, A&A, 572, A70 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rommel, J. B., Goumans, T. P., & Kästner, J. 2011, J. Chem. Theory Comp., 7, 690 [CrossRef] [Google Scholar]
- Santra, G., Sylvetsky, N., & Martin, J. M. L. 2019, JPCA, 123, 5129 [NASA ADS] [CrossRef] [Google Scholar]
- Schreiner, P. R., Reisenauer, H. P., Ley, D., et al. 2011, Science, 332, 1300 [NASA ADS] [CrossRef] [Google Scholar]
- Scibelli, S., Shirley, Y., Vasyunin, A., & Launhardt, R. 2021, MNRAS, 504, 5754 [NASA ADS] [CrossRef] [Google Scholar]
- Senevirathne, B., Andersson, S., Dulieu, F., & Nyman, G. 2017, Mol. Astro., 6, 59 [NASA ADS] [Google Scholar]
- Shingledecker, C. N., Tennis, J., Gal, R. L., & Herbst, E. 2018, ApJ, 861, 20 [NASA ADS] [CrossRef] [Google Scholar]
- Simons, M. A., Lamberts, T., & Cuppen, H. M. 2020, A&A, 634, A52 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Song, L., & Kästner, J. 2017, ApJ, 850, 118 [NASA ADS] [CrossRef] [Google Scholar]
- Truong, T. N., & Stefanovich, E. V. 1995, Chem. Phys. Lett., 240, 253 [NASA ADS] [CrossRef] [Google Scholar]
- Vazart, F., Ceccarelli, C., Balucani, N., Bianchi, E., & Skouteris, D. 2020, MNRAS, 499, 5547 [Google Scholar]
- Walsh, C., Millar, T. J., Nomura, H., et al. 2014, A&A, 563, A33 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Watanabe, N., & Kouchi, A. 2002, ApJ, 571, L173 [Google Scholar]
- Watanabe, N., Nagaoka, A., Hidaka, H., et al. 2006, P&SS, 54, 1107 [NASA ADS] [CrossRef] [Google Scholar]
- Woon, D. E., & Dunning, T. H. 1994, JCP, 100, 2975 [Google Scholar]
In this context, “hydrogenation” refers broadly to reactions involving H atoms, with distinctions between H abstraction and H addition noted where relevant.
Details on how to run the rev-DSD-PBEP86(D4) calculations with different codes can be found at https://www.compchem.me/revdsd-pbep86-functional
Cartesian coordinates supporting the calculations can be found at https://zenodo.org/records/14278652
In addition to our considerations, the numerical values for the rate constants are gathered in https://zenodo.org/records/14278652
All Tables
Reaction enthalpies (∆HR), activation energies (∆H‡), the absolute value of the imaginary frequency of vibration (Ω), and the crossover temperature (Tc) for the reactions of CH3CHO with H.
All Figures
|
Fig. 1 Acetaldehyde molecule with the positions where the hydrogenation and deuteration reactions were sampled and Newman projection of the molecular model showing the considered CH3CHO syn conformer. |
| In the text | |
|
Fig. 2 IRC profiles for Reactions (R1)–(R4) (We omit reaction (R5) due to the high ∆H‡). IRC profiles are presented at the rev-DSD- PBEP86(D4)/jun-cc-pV(T+d)Z not corrected by ZPVE, double-level CCSD(T), or implicit water ice environment (CPCM). |
| In the text | |
|
Fig. 3 Arrhenius plot of the hydrogenation rate constants (kH) for Reactions (R1)–(R4) including (dashed lines) and not including (solid lines) an implicit solvation model. All rate constants have been corrected for the implicit surface. Rate constants are presented until the lowest temperature achievable for each instanton in the sequential cooling scheme. |
| In the text | |
|
Fig. 4 Similar to Fig. 3 but including deuterium as an incoming particle (kD) for Reactions (R6) and (R7) |
| In the text | |
|
Fig. 5 Potential energy scan for Reactions (R8) (blue curve), (R9) (red curve), and (R10) (green curve) at the revDSD-PBEP86(D4)/jun-cc- pV(T+d)z level using a broken symmetry formalism. The discontinuities in r(H-H3) and r(H-O4) are caused by the insufficient resolution of the PES scan. The small bump in r(H-O4) is attributed to the constraints in the C-H bonds required to carry out the scan (see text). |
| In the text | |
|
Fig. 6 Schematic view of the CH3CO + H reaction. Depending on the initial configuration of the H atoms, the different kR must compete with diffusion, kDiff. If kR ≫ kDiff, then the reaction would dominate even in the presence of a barrierless channel, as is shown in the figure. BL means barrierless. |
| In the text | |
|
Fig. 7 Fourier-transform infrared profile of CH3CHO on c-ASW at 10 K. |
| In the text | |
|
Fig. 8 (a) Variation in the difference spectra of the solid CH3CHO after exposure to H atoms for 3, 60, and 120 min at 10 K. (b) Relative abundance of CH3CHO following a 2-hour exposure to H atoms (blue circles) compared to that with H2 molecules (red squares) on c-ASW at 10 K. |
| In the text | |
|
Fig. 9 TPD-QMS profile of pre-deposited CH3CHO with H atoms (black line) comparable with H2 molecules (blue line) on c-ASW at 10 K: m/z = 16 (top panel), m/z = 28 (middle panel), and m/z = 46 (bottom panel). |
| In the text | |
|
Fig. 10 FTIR spectrum of codeposition of CH3CHO and H atoms on Al at 10 K. |
| In the text | |
|
Fig. 11 TPD-QMS profile for the m/z = 14 and 42. Black arrows correspond to features attributable to H2CCO, obtained after the reaction of the pre-deposited CH3CHO with H atoms on c-ASW at 10K. The desorption peaks at 100–130 K and 140–150 K are derived from the remaining CH3CHO on c-ASW after exposure to H atoms. The peaks at 75–90 K and 150–165 K represent the desorption of H2CCO (Maity et al. 2015; Chuang et al. 2020; Fedoseev et al. 2022). |
| In the text | |
|
Fig. 12 Variation in the difference spectra of the solid CH3 CHO on the Al substrate at 10 K after exposure to D atoms for 10, 60, and 120 min comparable with the initial CH3CHO. |
| In the text | |
|
Fig. 13 TPD-QMS profile of pre-deposition of CH3 CHO with D atoms (black line) comparable with D2 (blue line) on Al substrate at 10 K, m/z = 30 (top panel); m/z = 17 (middle panel), and m/z =18 (bottom panel). The m/z = 30 (top panel) in the blue line represents the fragmentation of CH3CHO. |
| In the text | |
|
Fig. 14 TPD-QMS profile of m/z = 36 observed for the pre-deposition of CH3 CHO and D atoms (black line) is compared with that of D2 molecules (blue line) on the A1 substrate. |
| In the text | |
|
Fig. 15 TPD-QMS profiles of m/z = 49 and 50 (black lines) desorbed at around 130–200 K, likely corresponding to ethanol isotopologs CH3CD2OD and/or CH2DCD2OD. These species were yielded through the reaction of CH3CHO and D atoms, with behavior comparable to that of D2 molecules (blue line) on Al substrate at 10 K. |
| In the text | |
|
Fig. 16 TPD-QMS profile for the desorption peak observed at m/z = 48 (black line) may be attributed to the formation of CH3CHDOD through the addition reaction of D to CH3CHO. This is in the comparison to the desorption behavior for D2 (blue line) on the Al substrate at 10 K. |
| In the text | |
 |
Fig. A.1 Same as Fig. 2 but including reactions using an explicit 2 H2O model (dashed line). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.