| Issue |
A&A
Volume 687, July 2024
|
|
|---|---|---|
| Article Number | A230 | |
| Number of page(s) | 17 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202449378 | |
| Published online | 18 July 2024 | |
Single-atom catalysis in space
II. Ketene–acetaldehyde–ethanol and methane synthesis via Fischer-Tropsch chain growth★
1
Departament de Química, Universitat Autònoma de Barcelona,
08193
Bellaterra,
Catalonia,
Spain
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
2
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University,
Edinburgh,
EH14 4AS,
Scotland
Received:
29
January
2024
Accepted:
27
April
2024
Abstract
Context. The presence of grains is key to the synthesis of molecules in the interstellar medium that cannot form in the gas phase due to its low density and temperature conditions. In these reactions, the role of the grains is to enhance the encounter rate of the reactive species on their surfaces and to dissipate the energy excess of largely exothermic reactions, but less is known about their role as chemical catalysts; namely, bodies that provide low activation energy pathways with enhanced reaction rates. Different refractory materials with catalytic properties, such as those containing space-abundant d-block transition metals like iron (Fe), are present in astrophysical environments.
Aims. Here, we report for first time mechanistic insights into the Fischer-Tropsch-type (FTT) synthesis of ethanol (CH3CH2OH), through ketene (CH2CO) and acetaldehyde (CH3CHO) intermediates, and methane (CH4) via a chain growing mechanism using a single-Fe atom supported on silica (SiO2) surfaces as a heterogeneous astrocatalyst.
Methods. Quantum chemical simulations based on extended periodic surfaces were carried out to characterize the potential energy surfaces of the FTT chain growing mechanism. Calculations of the binding energies of reaction intermediates and products and Rice–Ramsperger–Kassel–Marcus kinetic calculations were performed to evaluate catalytic efficiencies and determine the feasibility of the reactions in different astrophysical environments.
Results. Mechanistic studies demonstrate that the FTT chain growing mechanism enters into direct competition with FTT methanol formation, since formation of the CH2 chain growth initiator is feasible. The coupling of the CH2 with CO (forming ketene) and subsequent H2 additions yield acetaldehyde and finally ethanol, while direct H2 addition to CH2 produces methane. Thermodynamically, both processes are largely exergonic, but they present energy barriers that require external energy inputs to be overcome. Kinetic calculations demonstrate the strong temperature dependency of the FTT processes as tunneling does not dominate.
Conclusions. The results could explain the presence of CH3CH2OH and CH4 in diverse astrophysical regions where current models fail to reproduce their observational quantities. The evidence that the chain growing mechanism is operating opens a new reactivity paradigm toward the formation of complex organic molecules, which is constrained by the temperature-dependent behaviour of the FTT reactions and by making their energy features a crucial aspect.
Key words: astrochemistry / molecular processes / solid state: refractory / stars: formation / ISM: molecules
The data underlying this article are freely available in Zenodo at https://doi.org/10.5281/zenodo.10556610
© The Authors 2024
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
The formation of complex organic molecules in the interstellar medium (ISM) is central to chemical evolution in space, which can ultimately be linked to the origin of life. Interstellar complex organic molecules, also known as iCOMs (Ceccarelli et al. 2017; Herbst 2017; Herbst & Van Dishoeck 2009; Ceccarelli 2023), were first observed in massive star formation regions (Rubin et al. 1971). However, it was not until their detection in regions that will eventually form Solar-like planetary systems that a direct link was made between organic chemistry in the ISM and that in the solar system (Caselli et al. 2003; Cazaux et al. 2003), supporting the idea of the “universal chemical seeds of life” proposed by De Duve (De Duve 2005, 2011).
The importance of iCOMs resides in their chemical structures. Although they are relatively small by terrestrial standards (between 6 and 13 atoms), they contain carbon (hence rendering the organic character) in combination with hydrogen, nitrogen, and/or oxygen (forming, for instance, formamide (NH2CHO), the simplest iCOM containing these four fundamental atoms for life; Saladino et al. 2012). Chemical evolution of iCOMs gives rise to other compounds of enhanced complexity that can be of prebiotic interest, such as amino acids, fatty acids, sugars, and nucleobases, which are indeed biomolecular building blocks for terrestrial life (Sandford et al. 2006; Altwegg et al. 2016; Pizzarello 2006). Given that iCOMs represent the dawn of organic chemistry, understanding the synthesis of iCOMs is currently a hot topic in astrochemistry.
Although in the spotlight, the routes of the formation and destruction of iCOMs are still widely debated (Caselli & Ceccarelli 2012; Vasyunin & Herbst 2013; Enrique-Romero et al. 2016; Vasyunin et al. 2017; Balucani et al. 2015). One of the prevailing paradigms postulates that they form on the surfaces of icy interstellar grains. The proposed mechanism suggests that hydrogenated frozen molecules (as components of the ices) can be broken by photolysis and radiolysis, giving rise to radicals, which in turn can couple, thereby forming iCOMs. When the ice sublimates, the newly formed iCOMs are injected into the gas phase, where they can be observed via their rotational lines (Garrod et al. 2008). Although radical coupling reactions have helped astrochemists rationalize the formation of simple, fundamental molecules such as H2 and NH3 (Ferrero et al. 2023a; Pantaleone et al. 2021), these reactions have some weak points regarding the formation of iCOMs: (i) they can present energy barriers, when they were initially advocated to be barrierless, (ii) they can present competitive reactions, such as the H abstraction from one radical to the other, and (iii) they can only occur in a particular temperature window, between the diffusion of the radicals and their sublimation (Enrique-Romero et al. 2021, 2022). Accordingly, alternative mechanisms alleviating some of these drawbacks have been postulated (Jin & Garrod 2020; Perrero et al. 2022). However, all of them are based on a grain-surface chemistry occurring on ice mantles, and accordingly they are only valid in the early stages of the planetary systems formation (e.g., in dark and/or prestellar cores), in which temperatures are cold enough to form and conserve the ices. However, iCOMs have also been detected (or postulated to be present) in other evolutionary stages in which ices are less dominant, like proto-stellar environments (e.g., hot cores or corinos; Ceccarelli et al. 2017; Bianchi et al. 2019, 2020) and protoplanetary disks (Walsh et al. 2014; Öberg et al. 2023). Additionally, there is increasing evidence that ices do not fully cover the grain cores due to their (likely) porous or even fractal nature (Potapov et al. 2020) in part due to space weathering of the grains (Bennett et al. 2013), in such a way that part of the refractory material is exposed to the gas-phase environment (Marchione et al. 2019). Thus, even in deep cold interstellar environments, synthesis of iCOMs can be enhanced by the potential catalytic properties of the refractory component of the dust grains.
Bare interstellar grains consist of refractory materials, among which Mg/Fe-silicates (usually in an amorphous state) are the most abundant (Duley 2000; Keller et al. 2002; Sargent et al. 2009). The underlying idea of the grain-surface chemistry is that some chemical reactions are favored when they occur on grain surfaces, in particular where gas-phase routes are deemed to be inefficient. In this sense, interstellar grains are postulated to act as reactant concentrators, third bodies, and/or chemical catalysts. While in interstellar icy grains clear evidence has been shown of the two former roles (Enrique-Romero et al. 2016, 2019, 2022; Pantaleone et al. 2021; Ferrero et al. 2023a,b; Fredon et al. 2021; Molpeceres et al. 2023), their capability as true chemical catalysts (namely, species that provide alternative pathways with lower activation energy barriers) is more limited (Lodders & Fegley 2023).
However, as was mentioned above, beyond ices there are other materials (refractories) that can present catalytic properties. Observations prove the presence of transition metals (i.e., elements belonging to the d-block of the periodic table) in the ISM, of which iron (Fe) is the most abundant. Interestingly, while Fe atoms naturally comprise part of the Fe-containing silicate structure, they can also be deposited on the grains from the gas phase, giving rise to single-Fe atoms supported on the grain surfaces (Sasaki et al. 2001; Nuth III et al. 2006).
Although very scarce, research on astrocatalysis of refractory materials is not unreported. Recent studies have focused on the occurrence of Fischer-Tropsch-type (FTT) reactions leading to the formation of alcohols and short-chain alkenes. The FTT reactions have been the object of study in astrochemistry because their reactants (H2 and CO) are the most abundant gas-phase molecules in the ISM. Dedicated laboratory experiments have successfully synthesized hydrocarbons using transition metal-containing dust analogues under simulated solar nebula conditions (Llorca & Casanova 1998; Ferrante et al. 2000; Sekine et al. 2006; Cabedo et al. 2021). These experiments demonstrated that true catalysis on grains can indeed occur. However, there is a lack of comprehension of the mechanistic steps involved, and the related energetics and kinetics of these FTT processes, which is fundamental to understanding the feasibility and operating conditions of these reactions in astrophysical environments.
In a very recent work (Pareras et al. 2023), we investigated by means of quantum chemical simulations and kinetic calculations the synthesis of methanol (CH3OH) through FTT reactions catalyzed by Fe single-atoms supported on silica (SiO2) surfaces, which is formed by successive H2 additions to CO. Our results demonstrate that, for the process to be feasible, a source of energy is required to overcome the associated activation energies forming CH3OH, a condition that can be achieved in higher-temperature astrophysical environments more easily than in deeply cold dense ones. Nevertheless, FTT reactivity is a complex process that goes beyond the synthesis of methanol, in which larger molecules can also be obtained following a chain growth mechanism. In fact, the mechanism of Fischer-Tropsch synthesis, or more accurately the chain growing mechanism that leads to the formation of alkanes, alkenes, alcohols, and the long-chain derivatives, is still under debate since the original paper by Fischer and Tropsch (Fischer & Tropsch 1923, 1926; Schulz 1999; Van Der Laan & Beenackers 1999; Khodakov et al. 2007; De Smit & Weckhuysen 2008; Cheng et al. 2010; Li et al. 2011; Psarras & Ball 2015; Foppa et al. 2018).
In the present work, we focus on the synthesis of ethanol (CH3CH2OH) through a chain growth mechanism. The key point differing this from FTT-CH3OH synthesis is that the adsorption of CO and H2 leads to the activation and dissociation of CO, forming CH2, which acts as the chain initiator. Ethanol is then obtained by the coupling between the CH2 and a second CO molecule (forming first ketene, CH2CO), followed by subsequent hydrogenations with H2, through acetaldehyde (CH3CHO) formation as an intermediate. However, as will be seen, the first steps of the chain growth are based on the H2 reactivity with CO, and accordingly it is in direct competition with the CH3OH formation. Thus, a comparison between the two processes is presented. Additionally, H2 addition to the CH2 initiator can also give rise to CH4, becoming a potentially competitive reaction to ethanol formation, and accordingly it has also been studied here. For the sake of clarity, all of these processes are schematically sketched in Fig. 1. Moreover, we carry out kinetic calculations to assess the catalytic performance of the Fe single-atom astrocat-alyst model in the investigated reactions, as well as to determine those favorable conditions at which the reactions can occur in agreement with the different astrophysical environments.
Due to our experience in FTT-CH3OH formation, where it was shown that Fe0 was better than the Fe2+ as single-atom catalyst on SiO2 surfaces, here reactivity is only addressed on the former system, which is hereafter referred to as Fe0@SiO2.
2 Methodology
The Fe0@SiO2 surface catalyst was modeled adopting a periodic approach (more details on how they were constructed and their structure are provided below). Geometry optimizations to characterize the potential energy surfaces (PESs) were carried out with the CP2K package (Kühne et al. 2020). The semi-local Perdew-Burke-Ernzerhof (PBEsol) functional was adopted (Perdew et al. 2008) together with the Grimme D3(BJ) correction to include dispersion forces (Grimme et al. 2010), combined with a double-ξ basis set (DZVP-MOLOPT-SR-GTH Gaussian basis set) for all the atom types, and a cutoff energy, Ecut = 500 Ry, for the plane wave auxiliary basis set. Core electrons were described with the Goedecker–Teter–Hutter pseu-dopotentials (Goedecker & Teter 1996) and valence ones with a mixed Gaussian and plane waves (GPWs; Lippert et al. 1997). Due to the size of the unit cell (see below), CP2K calculations were performed at the Γ point.
To refine the energies of the stationary points, single point calculations on the PBEsol-optimized geometries were performed employing B3LYP (a hybrid generalized gradient approximation (GGA) functional of Becke-Lee, Parr, and Yang; Lee et al. 1988; Becke 1993; Stephens et al. 1994), including the Grimme’s D3(BJ) correction term (Grimme et al. 2010), and using the triple-ξ basis set (TZVP) with the ab initio CRYS-TAL17 code (Dovesi et al. 2018), which adopts Gaussian-type orbitals (GTOs) as basis sets. The threshold parameters for the evaluation of the Coulomb and exchange bi-electronic integrals (the TOLINTEG keyword in the CRYSTAL17 code; Dovesi et al. 2018) were set equal to 7, 7, 7, 7, and 14, and calculations were also performed at the Γ point. In our previous work (Pareras et al. 2023), we already assessed that changing codes (namely, CP2K for geometry optimizations and CRYSTAL17 for single-point energy calculations) has a very low impact on the basis set employed (GPWs and GTOs, respectively).
Transition states structures were searched using the climbing image nudged elastic band (CI-NEB) technique implemented in CP2K (Kühne et al. 2020). Activation energy barriers were calculated as
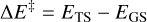 (1)
(1)
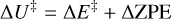 (2)
(2)
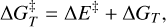 (3)
(3)
where ΔE‡ is the potential energy barrier, in which ETS and EGS are the absolute potential energies for the transition state and the local minimum structure, respectively, ΔU‡ is the vibrational zero-point energy (ZPE) corrected barrier, in which ΔZPE refers to the contribution of the ZPE corrections to ΔE‡, and 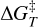 the Gibbs activation barrier at a given temperature, in which ΔGT refers to the contribution of the Gibbs corrections to ΔE‡.
the Gibbs activation barrier at a given temperature, in which ΔGT refers to the contribution of the Gibbs corrections to ΔE‡.
Analogously, reaction energies are defined as ΔErx, ΔUrx, and ΔGTrx as the reaction energies from the potential energies, including the ZPE corrections and the Gibbs reaction energies at a given temperature, respectively.
Binding energies (BEs) were computed using the counterpoise method as implemented in CRYSTAL17 in order to avoid the basis set superposition error (BSSE; Dovesi et al. 2018). Final BEs were calculated as
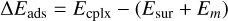 (4)
(4)
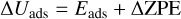 (5)
(5)
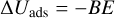 (6)
(6)
where ΔEads is the potential adsorption energy, Ecplx, Esur, and Em are the absolute potential energies for the adsorption complex, the isolated surface, and the isolated molecule, respectively, ΔUads is the ZPE-corrected adsorption energy (in which ΔZPE refers to the contribution of the ZPE corrections to ΔEads), and BE is the binding energy (which is ΔUads in opposite sign).
Frequency calculations were performed to validate the nature of the stationary points (i.e., minima with only real frequencies and transition states with one, and only one, imaginary frequency leading to the corresponding product). Vibrational harmonic frequencies were calculated at the PBEsol-D3BJ/DZVP-optimized structures using the finite differences method as it is implemented in the CP2K code. A partial Hessian approach was used to reduce the computational cost of the calculations. Thus, the vibrational frequencies were calculated for only a fragment of the entire system, which included the Fe metal center, the reactive species and the closest SiO2 moieties of the silica surface The atomic zone considered in the partial Hessian marrix calculation is highlighted in Fig. 2 (dashed line box).
The catalytic performance of the simulated FTT processes was investigated through reaction rate kinetic calculations. To this aim, a rate constant associated with each elementary bar-rier was calaulated using the Rice-Ramsperger-Kassel-Marcus (RRKM) theory (Marcus 1952), a microcanonical approximation that assumes that the phase space is statistically populated. In this RRKM treatment, tunneling effects were taken into account by adopting the unsymmetrical Eckart potential barrier model (Eckart 1930). For the calculation of the rate constants, we used the calculated vibrational freouencies as degrees of free-dom in the sum of states. Although we calculated a partial Hessian matrix to derive a set of vibrational modes, they are those directly involved in the reaction, and accordingly those that have a direct impact on the rate constants. The rest of the vibrational modes, which are not accounted for, belong to the surface inner layers and are assumed to have a negligible influence in the chemical reactions and by extension the rate con-stants (Molpeceres et al. 2023). These kinetic calculations were performed with a homemade program in which the RRKM algorithms were implemented for grain-surface processes, and are freely available in Enrique-Romero & Rimola (2024).
The Fe0@SiO2 catalytic system was built up by adding on an amorphous SiO2 periodic surface a single atom of Fe in its ground triplet state (see Fig. 2). The amoophous SiO2 surface was taken from the work of Ugliengo et al. (2008), in which the Fe0 center was added on a low density silanol (SiOH) silica surface (1.5 SiOH nm−2). It contains 187 atoms per unit cell, with cell parameters of a = 12.186 Å, b = 12.772 Å, and c = 49.317 Å, and α = 90.00◦, β = 90.00◦, and γ = 90.25◦, and with a thickness of 16 Å, leaving an empty space of 30 Å between surface replicas in the z direction (see Fig. A.1). A more detailed description of the catalyst construction can be found in our previous published article (Pareras et al. 2023).
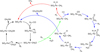 |
Fig. 1 Schematic representation of the FTT processes simulated in this work. Green line: C-O dissociation of acetaldehyde (CH2O), forming the CH2 chain initiator and O, and H2 addition to atomic O to form H2O. Blue lines: formation of ethanol (CH3CH2OH) by (i) reaction of CH2 with CO, forming ketene (CH2CO), and (ii) two H2 additions to ketene to form first acetaldehyde (CH3CHO) and finally ethanol. Red lines: formation of methane (CH4) by H2 addition to CH2. |
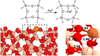 |
Fig. 2 Top: scheme of the Fe0 grafting on the SiO2 surface. Bottom: zoom-out (left) and zoom-in (right) of the optimized geometry of the Fe0 @SiO2 system. The dashed line box represents the atoms included in the partial Hessian matrix calculation. Relevant distances are depicted in Angstroms (Å). Color-coding: H atoms are represented in white, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
3 Results
The different reactions considered in the following subsections are depicted in Table 1, and in Figs. 3 and 4. The nomenclature used to label the minima of the PESs refer to the different moieties the are coordinated with the Fe center, where the atoms establishing the coordination with the Fe center are highlighted with the symbol (*). For instance, the local minima labelled as [*H2+ *CO] refers to the Fe0@SiO2 system with the H2 and CO moieties coordinated with the Fe center. The nomenclature adopted for the saddle points is different and depends on the type of process. For the reactions involving chemical bond cleavages (Fig. 3), the nomenclature of the saddle points specifies the moiety that is breaking. For instance, TSC–O is the saddle point for the breaking of the CO moiety into C + O, and TSHC–O is the saddle point for the breaking of the HCO moiety into HC + O. For reactions involving the ethanol and methane formation, the saddle points are named in order of appearance (e.g., TS1, TS2, TS3, etc.) following the sequence of reactions shown in Fig. 4. For the formation ethanol, the saddle points cover the TS1 TS5 range (see Fig. 4A), while the formation of methane involves TS6 and TS7 (see Fig. 4B).
Relative energies of the CO bond dissociation and H2O formation.
3.1 Toward the chain growth mechanism: CO dissociation and CH2 + H2O formation
Before delving into the chain growth mechanism it is important to understand how a chain initiation can be achieved. One possible route is based on the surface carbide mechanism, the first and oldest mechanism proposed for the FT synthesis (Fischer & Tropsch 1923, 1926; Biloen et al. 1979; Dictor & Bell 1986). This mechanism presumes dissociative adsorption of CO and H2 followed by the formation CH2 which combines and inserts into growing chains. Since we are dealing only with a single-Fe atom, it is likely that the CO molecule adsorbs first on the Fe center and posteriorly dissociates through an energetic barrier. It should be noted that the activation or dissociation of CO is in direct competition with its hydrogenation from H2, forming methanol. Therefore, as was mentioned earlier, energetic values obtained here are compared to those for the CH3OH formation (Pareras et al. 2023).
Following the proposed mechanism of entry 1 of Table 1, the ZPE-corrected PES for the CO dissociation was calculated and compared to that for the methanol formation (Fig. 3). The barrier needed to overcome the CO dissociation is extremely high (ΔU‡(C-O) = 95.2 kcal mol–1). Moreover, the process is highly endoenergetic (ΔUrx(C-O) = 26.4 kcal mol–1). Therefore, this surface carbide mechanism is energetically restricted.
However, recent works also demonstrate that the energy required for the CO dissociation can be reduced upon its hydrogenation. That is, the CO becomes activated due to being hydrogenated, in which the possible resulting product dissociates through the C-O bond (reassembling in part the two other main chain growth mechanisms, the surface enol mechanism (Kummer & Emmett 1953; Vannice 1976; Huff & Satterfield 1984) and the CO insertion mechanism (Henrici-Olivé & Olivé 1976; Masters 1979)). Here, we consider this CO dissociation to be mediated by multiple hydrogena-tions from H2 molecules. Remarkably, in a previous work by us (Pareras et al. 2023), it was shown that the interaction of H2 with the Fe single-atom leads to a spontaneous H2 homolytic cleavage. Moreover, the migration of the resulting H atoms to the bare SiO2 surfaces (that is, leaving the Fe single-atom center) is unlikely due to the stronger BE of H on the Fe0@SiO2 center than the bare SiO2 (84.9 and 2.6 kcal mol–1, respectively). Thus, the CO hydrogenation for its activation is simulated from H atoms interacting with the Fe center, in which three different scenarios are possible upon CO hydrogenation: (i) dissociation of HCO into CH and O (entry 2 of Table 1); (ii) dissociation of CH2O into CH2 and O (entry 3 of Table 1); and (iii) dissociation of CH2OH into CH2 and OH (entries 4 and 5 of Table 1). Interestingly, in all three scenarios, after the C-O bond breaking, formation of CH2 plus H2O occurs, leaving as a chain initiator the CH2 entity. Again, all of them are in direct competition with the direct CO hydrogenations that lead to the CH3OH formation, and therefore their energetics are also compared. All the obtained results are shown in Fig. 3.
Our results do not differ from the literature, and hydrogena-tion of the CO molecule activates it, reducing the energy required to break the C-O bond. Even after the first hydrogenation, the required barrier is reduced by half with respect to direct CO dissociation (ΔU‡(HC-O) = 45.7 kcal mol–1), but the process is still endoenergetic (ΔUrx(HC-O) = 28.3 kcal mol–1). The energy required to break the CO molecule hits the lowest value after the second hydrogenation (namely, dissociation of H2CO into CH2 and O), reaching a barrier of ΔU‡(H2C-O) = 20.0 kcal mol–1, with an importantly reduced reaction energy of ΔUrx(H2C-O) = 1.1 kcal mol–1. A third hydrogenation is also evaluated, forming the CH2OH moiety prior to the CO dissociation. The hydrogenation of the oxygen atom (forming CHOH) has already been reported to be higher in energy than the C atom (forming CH2O; Van Der Laan & Beenackers 1999; Psarras & Ball 2015; Foppa et al. 2018, 2019; Li et al. 2011). Our results are in good agreement with the literature data (reported in Table B.1), with a barrier for the CH2OH and CH3O formation of 52.7 and 40.0 kcal mol–1, respectively. The instability of CH2OH, however, does not imply a reduction in the CO dissociation, as its barrier is ΔU‡(H2C-OH) = 35.0 kcal mol–1, but rather presents an exoenergetic character (ΔUrx(H2C-OH) = –23.3 kcal mol–1. Thus, these results indicate that the most energetically favorable path toward CO dissociation is to form first CH2O, followed by its H2C-O dissociation. This path, moreover, has the advantage that the dissociation products are O and CH2, this latter already being the chain growth initiator species in the form of the CH2-Fe0@SiO2 surface system.
As was mentioned above, and from the data collected in the energy profile of Fig. 3, the hydrogenation of CO to induce its dissociation is in direct competition with the CH3OH formation. By comparing the energy features of both processes (Fig. 3), direct CO dissociation and HC-O dissociation are more energetic steps than CO hydrogenation to reach CH2O. At this point, the reaction can advance toward either methanol formation (with an H2 adsorption and subsequent hydrogenation of CH2O, with intrinsic ΔU‡(CH3O) = 12.8 kcal mol–1 and ΔU‡(CH3OH) = 17.0 kcal mol–1, both steps being exoenergetic, ca. 6 kcal mol–1 each; see Fig. 3) or breaking the CH2O into CH2 and O (with ΔU‡(H2C-O) = 20.0 kcal mol–1), with the resulting O atom, according to the FT reactivity, needing to be hydrogenated to form H2O. Entries 4 and 5 of Table 1 collect the energetics for the two hydrogenations needed to form H2O (processes that are also represented in Fig. 3). The results show that the first and second hydrogenations have energy barriers of ΔU‡(O-H) = 23.5 kcal mol–1 and ΔU‡ (HO-H) = 12.0 kcal mol–1 and exoenergetic reaction energies of ΔUrx(O-H) = –15.3 kcal mol–1 and ΔUrx(HO-H) = –25.0 kcal mol–1. Accordingly, the results indicate that the formation of CH3OH and the formation of CH2 + H2O are in fair competition, since both processes present similar energy barriers (although CH3OH formation is energetically slightly favored) and are exoenergetic steps (the formation of CH2 + H2O presenting more negative reaction energy by some amount). Therefore, all the presented results relative to the CO dissociation and the formation of the CH2-Fe0@SiO2 chain initiator indicate that, from the mechanistic point of view, the processes are feasible.
The barriers needed to overcome the H2C-O bond breaking and form CH2 and O and the hydrogenation of O to form H2O are relatively energetically affordable.
 |
Fig. 3 (A) Reaction mechanism for the CO dissociation into C + O. (B) Reaction mechanism for the HCO dissociation into HC + O. (C) Reaction mechanism for the H2CO dissociation into H2C and O. (D) Reaction mechanism for the first oxygen hydrogenation to OH. (E) Reaction mechanism for the second oxygen hydrogenation to H2O. (F) ZPE-corrected PESs (in kcal mol–1) for the different proposed CO dissociation mechanisms, as well as of H2O and CH3OH formation, using as the 0th reference energy the Fe0@SiO2 + H2 + CO asymptote. The optimized geometries of the five transition state structures involved in the three different C-O dissociations and in the H2O formation are also shown (bond distances in Å). The asterisk symbol (*) denotes from which atom the relevant molecule is coordinated with Fe. Color-coding: H atoms are represented in white, C atoms in gray, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
 |
Fig. 4 (A) Reaction mechanism for the ethanol formation from the direct CH2 – CO coupling forming CH2 and O, the reaction of CH2 with an incoming CO, forming H2CCO, and subsequent hydrogenations. (B) Reaction mechanism for the methane formation from hydrogenation of CH2. (C) ZPE-corrected PESs (in kcal mol–1 ) for the ethanol formation (black) and methane formation (gray) mechanisms, using as the 0th reference energy the Fe0@SiO2-CH2 + H2 + CO asymptote. The optimized geometries of all the transition state structures (bond distances in Å) are also shown. The asterisk symbol (*) denotes from which atom the relevant molecule is coordinated with Fe. Color-coding: H atoms are represented in white, C atoms in gray, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
3.2 Ethanol formation
The reaction mechanism proposed for the ethanol formation from CH2-Fe@SiO2 is depicted in Fig. 4A, and the computed ZPE-corrected PES in Fig. 4C.
The first step is the favorable adsorption of a new CO molecule on the Fe center of the CH2-Fe0@SiO2 system, and the first barrier is the direct coupling between CH2 and CO, forming ketene (CH2CO). The energy barrier is relatively small (ΔU‡(TS1) =1.9 kcal mol–1), and the reaction step is largely exoenergetic (ΔUrx(TS1) = –20.7 kcal mol–1). These favorable energetics can be explained by considering the CH2 molecule as a Fischer-carbene system (see Appendix B and Fig. D.1). In terms of reactivity, these carbene types show a strong elec-trophilicity, prone to couple with the relatively nucleophile C atom of CO (Santamaría & Aguilar 2016; Epping et al. 2023).
The next step is the hydrogenation of ketene by reacting with a new H2 molecule to form acetaldehyde (CH3CHO). This starts with the favorable adsorption of an incoming H2 on the Fe center (which in this case remains in the form of H2, at variance with the H2 addition to CO; see above), and then the addition of the H atoms of H2 to ketene. The energetics associated with the breaking of the adsorbed H2 molecule and hydrogenation of CH2CO are ΔU‡(TS2) = 31.1 kcal mol–1 and an endoenergetic ΔUrx(CH2CHO) = 18.5 kcal mol–1. We only considered the hydrogenation of the central C atom (and not the terminal one), as it has been shown to be more favorable according to previous mechanistic studies based on FTT chain growing mechanisms (De Smit & Weckhuysen 2008; Zhuo et al. 2009; Cheng et al. 2010; Psarras & Ball 2015; we note that this is not the case for H addition to ketene on ices, in which the terminal C atom has been shown to be more favorable Ferrero et al. 2023a). Due to the instability of the CH2CHO intermediate, the subsequent hydrogen transfer on the terminal C atom shows a small energy barrier (ΔU‡(TS3) = 1.7 kcal mol–1), and the formation of acetaldehyde is largely exoenergetic (ΔUrxCH3(CHO) = –40.1 kcal mol–1).
Finally, to proceed with the ethanol formation, a second H2 adsorption on the Fe center is considered, which is again slightly exoenergetic. As was mentioned above, the hydrogena-tion of the carbon atoms is energetically more favorable than on the oxygen atoms. Therefore, the following hydrogenations lead first to the formation of the CH3CH2O intermediate, with an energy barrier of ΔU‡(TS4) = 22.8 kcal mol–1 and an exoenergetic ΔUrx(H3CH2O) = –11.0 kcal mol–1, in which the newly formed CH3CH2O intermediate is coordinated on the Fe center only by the oxygen atom. The last hydrogenation leading to the formation of CH3CH2OH shows an energy barrier of ΔU‡(TS5) = 15.6 kcal mol–1 and is also an exoenergetic step with ΔUrx(CH3CH2OH) = –8.8 kcal mol–1.
3.3 Methane formation
For CH3CH2OH formation, we considered the adsorption of a CO molecule and its reactivity with CH2. However, H2 is the most abundant molecule in the ISM, and accordingly an H2 molecule is likely to adsorb before a CO molecule, and hence enable the hydrogenation of CH2 to form CH4. Figure 4B shows this mechanism, and the calculated ZPE-corrected PES is also shown in Fig. 4C.
The reaction starts with the favorable adsorption of an H2 molecule. The first barrier concerns the breaking of the H2 molecule and the formation of the CH3 intermediate, which results in a relatively high activation barrier (ΔU‡(TS6) = 28.6 kcal mol–1), with a largely exoenergetic reaction energy (ΔUrx(CH3) = –56.6 kcal mol–1). Although the observed barrier is quite high, this data matches that of the previously observed barriers; that is, the first hydrogenations in the ethanol and methanol formation processes also present energy barriers above 20 kcal mol–1 , such that these are the rate-determining steps of respective processes. The second hydrogenation occurs through a very small barrier (ΔU‡ (TS7) = 1.2 kcal mol–1), finally leading to the exoenergetic formation of CH4 (ΔUrx(CH4) = –4.8 kcal mol–1).
Collected binding energies and relative energies.
4 Discussion
4.1 Binding energies of the formed species
The mechanistic study shown above gives an understanding of how the reactions evolve throughout the different reaction steps. Preliminary conclusions can be extracted from the computed energetics. An important aspect is that, in general, the stationary points are lower in energy than the asymptotes due to the highly favorable adsorption of the reactants; namely, the H2 and CO molecules on the Fe single-atom center. This means that the reactive system can have a surplus of energy at its disposal in the case in which the gained adsorption energy has not dissipated throughout the surface, which can accordingly be used to advance the reaction. That is, in the scenario in which all the transition states are below the asymptote, the reaction can progress in a barrierless fashion. However, this assumption has a possible drawback: if any of the intermediates has a BE below the asymptotes, that intermediate can easily be desorbed, hampering the evolution of the reaction. To provide deeper insight into that, we calculated the BEs of the different stable intermediates as well as of the final products on the Fe0 @SiO2 surface. These values are reported in Table 2.
The BEs of the H2 and CO reactants, as was shown in our previous work (Pareras et al. 2023), are relatively high as they chemisorb on the metal center. The BEs of the products (H2O, CH3CH2OH, and CH4) lie below their corresponding asymptotes. In contrast, the BEs of the intermediates (CH2CO, CH3CHO, and the CH2 chain initiator) are energetically above the asymptote. These values confirm that, in the scenario in which the energy has not dissipated across the surface, the reaction would advance, forming the desired products, as the intermediates are strongly coordinated with the metal center without the possibility of desorption. Moreover, in this same scenario, BE values also give some clues as to the formation of by-products. That is, in the studied paths, ketene and acetalde-hyde (reaction by-products) can be formed and retained on the metal center, and accordingly some amounts of these species could also be formed.
4.2 Kinetic analysis
In the previous section, we were considering a scenario in which the surplus of energy provided by the favorable chemisorption of the reactants has not dissipated, and is accordingly available and used for the reaction to progress. However, this scenario is a limiting case, difficult to take place as such, since it has been reported that solid state surfaces have energy dissipation capabilities, including not only ices (e.g., Pantaleone et al. 2021) but also metals (Rittmeyer et al. 2018). Thus, in this section, we deal with the opposite scenario: that in which the surplus energy of the exoenergetic elementary steps dissipates. In this scenario, the FTT reactions can be understood as a set of successive steps that need to be treated individually to know their feasibility according to their kinetics. Thus, here, kinetic calculations based on the RRKM theory (see the methods section above) were carried out to calculate rate constants (k) and derive Arrhenius plots for the most key elementary steps of the investigated processes (shown in Fig. 5). Kinetic analysis is useful to understand how each reaction elementary step behaves in terms of reaction rate variation with temperature and to evaluate the possibility of tunneling, allowing us to predict whether a step is favorable or not and under which environmental conditions (namely, the temperature). To this end, using the Arrhenius plots, we determined at which temperature the elementary steps have a k = 1 yr–1, considering this rate constant as the lower limit at which the reaction is relatively fast (according to astronomical timescales, taking the age of a molecular cloud as 106 yr).
In this work, three different possibilities to activate and dissociate CO and obtain the CH2 chain growth initiator have been proposed: (i) direct CO dissociation, (ii) single CO hydrogena-tion and dissociation of HC-O, and (iii) double CO hydrogena-tion and dissociation of H2C-O. In the two first cases, the energy barriers are higher than those leading to the H2CO and CH3OH formation. However, the energy barrier of the third possibility directly competes with the methanol formation. We calculated the rate constants for these three possibilities, and the results show that the first two do indeed need an important source of energy to proceed with the C-O and HC-O cleavages (Fig. E.1). In contrast, for the H2C-O dissociation, the kinetic results indicate that at a temperature of 205 K the rate constant is k = 1.19 yr–1 (Fig. 5A).
As is seen in the results section, the hydrogenation of the resulting species upon the H2C-O dissociation forming a H2O molecule is crucial to obtain the CH2 chain initiator (within the FTT framework). This hydrogenation involves two steps: (i) the dissociation of the H2 molecule and the first hydrogen transfer forming the OH intermediate, and (ii) the last hydrogen transfer leading to H2O. The RRKM calculations show that the first step has a rate constant of k = 1.07 yr–1 at a temperature of 255 K, in which tunneling effects do not dominate (as is seen in Plot A of Fig. E.2), but in the second step tunneling is relevant, giving rise to a rate constant of k = 1.10 yr–1 at a temperature of 15 K. These kinetic data reaffirm what was observed in the PES; that is, obtaining the CH2 chain initiator requires two energy demanding steps to be overcome, with the first one (the H2C-O breaking) being kinetically feasible only at relatively high temperatures (Fig. E.2).
The propagation mechanism toward ethanol formation starts with the coupling of CH2 and CO to form ketene. Interestingly, the energetics shown in Fig. 4 already show a very small barrier for this step (around 1 kcal mol–1). Kinetic calculations are in line with this barrier, in which, moreover, tunneling effects are predicted to be relevant (Fig. 5B), resulting in k = 1.28 yr–1 at T = 3 K, and accordingly one can consider this step to be feasible at any temperature. The next step concerns the hydro-genation of ketene forming acetaldehyde, which starts with the H2 dissociation and subsequent H addition, presenting a high energy barrier. Kinetic calculations agree with these energy features, resulting in the full dependence of k on temperature, in which k = 1.16 yr–1 is only reached at 318 K (Fig. 5C). According to these values, this is the rate-determining step of the whole FTT-ethanol synthesis. The second hydrogenation presents a very small energy barrier, with enhanced reaction rates and dominance of tunneling (k = 332.5 yr–1 at 5 K; see Fig. E.3).
The last two hydrogenations of acetaldehyde, which show lower energy barriers than the first hydrogenation of ketene, result in reasonable kinetics with a full temperature dependence of the first hydrogenation without any contribution from tunneling effects (k = 1.20 yr–1 and k = 1.08 yr–1 at T = 92 K, for the first and second hydrogenation, respectively; see Fig. E.3).
Finally, we also considered the path in which a carbene molecule is hydrogenated to form methane. As is seen above, the first hydrogenation steps involving the H2 breaking are the rate-determining steps of the mechanisms. Kinetic data of this process show a minor contribution from tunneling at very low temperatures, and accordingly the process can be considered to be purely dependent on the temperature (k = 1.05 yr–1 at T = 300 K; Fig. 5D). The second hydrogenation has a very small energy barrier, and the process is fully dominated by tunneling (Fig. E.4).
Interestingly, there are kinetic similarities between the FTT synthesis of CH3OH, CH3CH2OH, and CH4. The key step in all of them is the first hydrogenation via H2 breaking and the subsequent hydrogen transfer to CO, H2CCO, and CH2 (for CH3OH, CH3CH2OH, and CH4 formation mechanisms). However, in the CH3OH mechanism, this first barrier is slightly lower than the two other mechanisms due to the cooperative effect between a Si surface atom and the Fe center, which allows it to start with an already dissociated H2 molecule.
 |
Fig. 5 Arrhenius plots for the elementary steps associated with |
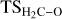
4.3 Astrophysical implications
One of the aims of this work is to determine if the syntheses of CH3CH2OH and CH4 via a FTT chain growing mechanism catalyzed by the Fe0 @SiO2 system are feasible within the astro-chemical framework, where the abundances of the reactants are very small, and the temperatures can vary from five to hundreds of Kelvins. Ethanol is deemed to be an important molecule for the formation of larger iCOMs (Skouteris et al. 2018), and has been detected toward a few different astrophysical environments. It has been seen in some star-forming regions: toward the continuum peak of some low-mass Class 0 protostars such as the binary IRAS 16293-2422 (Taquet et al. 2015) and NGC 1333-IRAS 4A (Taquet et al. 2015), where it is observed in the hot corino region. Notably, it has been detected in L1157-B1, where it is observed toward the protostellar shock region, showing an abundance at least two times larger than abundances in hot-corinos (Arce et al. 2008). It is usually detected toward intermediate-or high-mass star-forming regions, in the hot core region (Zuckerman et al. 1975; Millar et al. 1995; Ohishi et al. 1995; Belloche et al. 2009; Palau et al. 2011; Crockett et al. 2014; Rivilla et al. 2017), but its detection can remain relatively elusive when compared to other iCOMs (Blake et al. 1987; Friedel & Snyder 2008; Caux et al. 2011). Ethanol has been tentatively detected in ices at infrared (IR) wavelengths (Bisschop et al. 2008; Öberg et al. 2011; Boogert et al. 2015; McClure et al. 2023), but confirmed very recently (Rocha et al. 2024). Finally, ethanol has been detected multiple times in cometary bodies, such as Hale-Bopp, Lovejoy, Jupiter family comets, and Oort Cloud comets with abundances of ~0.1% wrt water (Biver & Bockelée-Morvan 2019).
The formation of ethanol in the ISM is still debated and poorly studied. Generally, studies suggest that it is formed on grain surfaces through photo-processing of methanol and water ices. Some models suggest that its production proceeds through the radical combination of CH3 and CH2OH (Garrod et al. 2008; Belloche et al. 2009; Rivilla et al. 2017). Others point to a non-energetic formation via a potentially barrierless interaction of CCH radicals with H2O ices to form vinyl alcohols, as precursors of ethanol (Perrero et al. 2022). Nevertheless, as was mentioned in the introduction, these types of mechanisms have the drawback of needing the presence of ices covering the dust grains, and hence can only apply to certain regions of the ISM; namely, those with temperatures low enough to ensure the existence of interstellar ices.
The formation of acetaldehyde (and hence the possible formation of ethanol by the latter’s hydrogenation) by the combination of HCO and CH3 is not possible in warm astrophysical environments because of the sublimation of the volatile icy species, leaving open the door for the formation of ethanol through pathways such as FTT synthesis. By contrast, in deep ultracold environments (where FTT reactions are not feasible), the formation of acetaldehyde by HCO and CH3 coupling can prevail, although computational studies question its occurrence (Balucani et al. 2015; Enrique-Romero et al. 2021, 2022). Our data indicate that, from both the energetic and kinetic points of view, the chain growing mechanism does indeed compete with the methanol formation, and both are viable at temperatures higher than 300 K. Therefore, enhancement of ethanol in hot or shocked regions, such as high-mass star-forming regions or stellar outflows, where temperatures can get high enough, is possible through Fe single-atom catalysis. However, it is true that CH3OH formation is relatively more favorable than chain growing, leading to a larger abundance when it is formed through this mechanism.
Experimentally, it has been shown that ethanol can be produced through Fe catalysis in reducing environments under different conditions of temperature and pressure (Cabedo et al. 2021). Under H and CO atmospheres, alcohol production, mainly methanol and ethanol, can be achieved using chondritic material as a catalytic support. However, the production of both alcohols only achieves 0.06 and 0.03% of the total CO consumed, respectively, at ~600–700 K, making it a small percentage of the total products. Those experiments show that methane production under those conditions is more favorable, starting at ~500 K and with production of up to 15%. Ethanol has also been experimentally formed, among other alcohols, using Fe meteoritic material and different silicates as support, under H and CO2 atmospheres (Peters et al. 2023). While the results are limited, both point to a possible synthesis of ethanol due to the presence of Fe particles in interplanetary material, under warmer temperature conditions, which could occur in many different environments like those mentioned above.
Moreover, ethanol has recently gained some attention as it has been touted as the precursor of several iCOMs formed by cold gas-phase reactions (Skouteris et al. 2018). The very first hydrogenations to CO are needed to form H2CO (as a previous step toward methanol) and to facilitate the dissociation of the H2C-O bond and give rise to the CH2 chain initiator. H2CO is a commonly observed molecule in the ISM, normally attributed to CO hydrogenation (Watanabe & Kouchi 2002; Roberts et al. 2004; Fuchs et al. 2009) or dust grain chemistry (Potapov et al. 2017; Inostroza-Pino et al. 2021). While our results show that H2CO would stay bonded to the catalyst, we could expect a certain amount of formation as a by-product of FTT chemistry. Ethanol is also usually observed accompanied by other molecules of which it is expected to be a parent molecule, such as glycoaldehyde or methyl and ethyl formate (Jørgensen et al. 2012; Rivilla et al. 2017). Additionally, the correlation of gly-colaldehyde and ethanol abundances observed toward several interstellar sources has been shown to closely follow the theoretical predictions when the synthesis of glycoaldehyde takes place through a gas-phase scheme after the desorption of ethanol from the dust grain surfaces (Vazart et al. 2020).
Finally, it is worth mentioning that, in the considered mechanisms, CH2 forms in situ as an intermediate species of the FTT synthesis, but one should also bear in mind that CH2 has been observed in the ISM, with a first detection by Hol-lis et al. in 1988 (Hollis et al. 1989) that was confirmed later in 1995 (Hollis et al. 1995). Thus, CH2 is an available species that makes suitable the scenario in which a gas-phase CH2 species strongly adsorbs on the single-atom Fe center. Accordingly, the investigated CH3CH2OH and CH4 formation paths also hold considering this possibility, irrespective of the competition between the CH3OH formation and the H2C-O dissociation.
5 Conclusions
In this work, the formation of ethanol (subsequent to the formation of ketene and acetaldehyde) and methane following a FTT chain growing mechanism catalyzed by Fe0 single-atoms supported on a silica (SiO2) surface has been investigated by means of quantum chemical simulations and kinetic calculations.
The main conclusions are:
- 1.
A plausible ethanol formation reaction mechanism, catalyzed by Fe0@SiO2, is presented from which the required chain initiator (CH2) for the chain growing mechanism is formed. The process involves C-O dissociation and the subsequent formation and desorption of H2O. The proposed mechanism shows different energetic barriers to overcome for the reaction to advance, and therefore the energetic requirements play a key role.
- 2.
Two possible mechanisms are reported whereby the reactant CH2 can be coupled with a CO molecule, leading to ethanol formation, or hydrogenated, forming CH4. The proposed reaction mechanisms also show different barriers that need to be overcome, making these processes always restricted to a source of energy.
- 3.
We have demonstrated that, in general, the chemical systems work below the asymptote due to the strong BE between the catalyst and the reactants. This fact is translated into a surplus of energy that, in the scenario in which this energy is not dissipated, the proposed processes would advance in a barrierless fashion. Within this scenario, calculated BEs show that the intermediates are strongly attached to the Fe metal center, while the desired products show lower BEs. This demonstrates that the reaction will not stop in the intermediates, as those will not desorb, preventing the reaction from advancing, always considering that the energy is not dissipated.
- 4.
In the scenario where the adsorption energies are fully dissipated throughout the surface, our simulations indicate that the formation of a chain initiator CH2 and the iCOMs CH3CH2OH and CH4 are, with energy requirements, affordable in different astrophysical environments.
Our results demonstrate that the formation of CH3CH2OH and CH4 catalyzed by the heterogeneous Fe0@SiO2 catalyst is operative in diverse astrophysical environments. Moreover, the possibility of generating a CH2 chain initiator makes this reaction process more versatile to the formation of different iCOMs beyond ethanol and methane. This opens up new and unexplored scenarios for the formation of even more complex C-containing molecules in space.
Acknowledgements
This project has received funding within the European Union’s Horizon 2020 research and innovation program from the European Research Council (ERC) for the project “Quantum Chemistry on Interstellar Grains” (QUANTUMGRAIN), grant agreement No 865657. G.P. thankfully acknowledges financial support by the Spanish Ministry of Universities and the European Union’s Next Generation EU fund for a Margarita Salas contract. Spanish MICINN is also acknowledged for funding the projects PID2021-126427NB-I00 and CNS2023-144902. The UK Engineering and Physical Science Research Council (EPSRC, EP/W023024/1) is also acknowledged. The authors thankfully acknowledge RES resources provided by Univ. Valencia for the use of Tirant (activity QHS-2022-2-0022) and by BSC for the use of MareNostrum (activity QHS-2023-1-0019). The supercomputational facilities provided by CSUC are also acknowledged. The EuroHPC Joint Undertaking through the Regular Access call project no. 2023R01-112, hosted by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID: 90254) is also acknowledged.
Appendix A Unit cell considered in this work
 |
Fig. A.1 Representation of the optimized unit cell considered in this work. |
Appendix B Literature reported energies for HCOH formation versus H2CO formation
Relative energies reported in the literature for the HCOH and H2CO formation.
Appendix C Energetic data for the synthesis of the CH2OH intermediate and the CH2-OH breaking
As was mentioned in the main text, it has been evaluated that the CH2OH intermediate forms from the hydrogenation of the O atom in the CH2O intermediate. This scenario was postulated in order to study the possible awakening of the C-O bond through the hydrogenation of the O atom. Energetic data shows that, apart from the fact that the hydrogenation of the O atom prior the saturation of the C atom in the CH2O intermediate is already less energetically favorable (the relevant energetic barrier is higher), this does not represent a reduction in the energy required for the C-O breaking. Table C.1 collects the relative energies (Rel Es) of the CH2O hydrogenation to form CH2OH and the H2C-OH breaking.
Relative energies for the the H2CO hydrogenation and H2C-OH bond dissociation.
Appendix D Carbene coordination with the Fe0@SiO2
The CH2 molecule is considered to be chemosorbed on the Fe center, showing a BE of 99.87 kcal mol–1 (we note that the ground state of the CH2 carbene in the gas phase has an electronic multiplicity of triplet (two unpaired electrons)). Although BE demonstrates the strong binding between the CH2 and the Fe center, the nature of the metal-carbene binding is a sensible topic in chemistry (Lee et al. 1988)). Here, we firstly assume that we are dealing with a Fischer-type carbene as this is usually featured in low oxidation state metals, mostly middle and late transition metals. Even if Fe0 is a first-row transition metal, here it is in its lowest oxidation state. In order to corroborate the true nature of the single-atom Fe0-Carbene interaction, a frontier molecular orbital study was performed. One of the con-sequences of a Fischer-carbene complex is a polarization toward the metal of the metal-carbene π-bond, resulting in a metal-ceniered π-HOMO and a carbon-centered π*-LUMO. Here, we are dealing with a triplet multiplicity; therefore, there will be two single occupied molecular orbitals (SOMOs). In Fig. D.1, the two SOMOs are plotted, as is the first LUMO. SOMO-1 corresponds to the σ-donation from the carbene ligand to the metal, while SOMO corresponds to the π-back donation from the metal to the ligand (in order to keep the same isovalue, π-orbitals of the carbene cannot be spotted in the latter one). Finally, the LUMO graphic shows a clear dominance of the π-orbitals of the carbene, affirming the carbon-centered π*-LUMO that was expected.
 |
Fig. D.1 Representation of the calculated frontier orbitals, from left to right: SOMO-1, SOMO, and LUMO. |
Appendix E Kinetic data
Appendix E.1: Arrhenius plots for the different C-O bond-breaking mechanisms
 |
Fig. E.1 Arrhenius plot for the free energy barrier associated with TSC–O (panel A), TSHC–O (panel B), TSH2C–O (panel C), TSCH2OH (panel D), and TSH2C–OH (panel E). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
Appendix E.2: Arrhenius plots for the H2O formation mechanism
 |
Fig. E.2 Arrhenius plot for the free energy barrier associated with TSO–H (panel A) and TSHO–H (panel B). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
Appendix E.3: Arrhenius plots for the CH3CH2OH formation mechanism
 |
Fig. E.3 Arrhenius plot for the free energy barrier associated with TS1 (panel A), TS2 (panel B), TS3 (panel C), TS4 (panel D), and TS5 (panel E). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
Appendix E.4: Arrhenius plots for the CH4 formation mechanism
 |
Fig. E.4 Arrhenius plot for the free energy barrier associated with TS6 (panel A) and TS7 (panel B). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1 . Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
References
- Altwegg, K., Balsiger, H., Bar-Nun, A., et al. 2016, Sci. Adv., 2, e1600285 [NASA ADS] [CrossRef] [Google Scholar]
- Arce, H. G., Santiago-García, J., Jørgensen, J. K., Tafalla, M., & Bachiller, R. 2008, ApJ, 681, L21 [NASA ADS] [CrossRef] [Google Scholar]
- Balucani, N., Ceccarelli, C., & Taquet, V. 2015, MNRAS, 449, L16 [Google Scholar]
- Becke, A. D. 1993, J. Chem. Phys., 98, 5648 [Google Scholar]
- Belloche, A., Garrod, R. T., Müller, H. S., et al. 2009, A&A, 499, 215 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bennett, C. J., Pirim, C., & Orlando, T. M. 2013, Chem. Rev., 113, 9086 [NASA ADS] [CrossRef] [Google Scholar]
- Bianchi, E., Codella, C., Ceccarelli, C., et al. 2019, MNRAS, 483, 1850 [Google Scholar]
- Bianchi, E., Chandler, C. J., Ceccarelli, C., et al. 2020, MNRAS, 498, L87 [NASA ADS] [CrossRef] [Google Scholar]
- Biloen, P., Helle, J. N., & Sachtler, W. M. H. 1979, Chem. Inf.-Dienst, 10, 95 [Google Scholar]
- Bisschop, S. E., Jørgensen, J. K., Bourke, T. L., Bottinelli, S., & Van Dishoeck, E. F. 2008, A&A, 488, 959 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Biver, N., & Bockelée-Morvan, D. 2019, ACS Earth Space Chem., 3, 1550 [Google Scholar]
- Blake, G. A., Sutton, E. C., Masson, C. R., & Phillips, T. G. 1987, ApJ, 315, 621 [Google Scholar]
- Boogert, A. C., Gerakines, P. A., & Whittet, D. C. 2015, ARA&A, 53, 541 [Google Scholar]
- Cabedo, V., Llorca, J., Trigo-Rodriguez, J. M., & Rimola, A. 2021, A&A, 650, A160 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Caselli, P., & Ceccarelli, C. 2012, A&Arv, 20, 1 [CrossRef] [Google Scholar]
- Caselli, P., Van Der Tak, F. F., Ceccarelli, C., & Bacmann, A. 2003, A&A, 403, 37 [Google Scholar]
- Caux, E., Kahane, C., Castets, A., et al. 2011, A&A, 532, A23 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cazaux, S., Tielens, A. G. G. M., Ceccarelli, C., et al. 2003, ApJ, 593, L51 [CrossRef] [Google Scholar]
- Ceccarelli, C. 2023, Faraday Discuss., 245, 11 [NASA ADS] [CrossRef] [Google Scholar]
- Ceccarelli, C., Caselli, P., Fontani, F., et al. 2017, ApJ, 850, 176 [NASA ADS] [CrossRef] [Google Scholar]
- Cheng, J., Hu, P., Ellis, P., et al. 2010, Top. Catal., 53, 326 [CrossRef] [Google Scholar]
- Crockett, N. R., Bergin, E. A., Neill, J. L., et al. 2014, ApJ, 787, 112 [Google Scholar]
- De Duve, C. 2005, Singularities: Landmarks on the Pathways of Life (Cambridge: Cambridge University Press), 1 [Google Scholar]
- De Duve, C. 2011, Philos. Trans. R. Soc. A, 369, 620 [NASA ADS] [CrossRef] [Google Scholar]
- De Smit, E., & Weckhuysen, B. M. 2008, Chem. Soc. Rev., 37, 2758 [CrossRef] [Google Scholar]
- Dictor, R. A., & Bell, A. T. 1986, J. Catal., 97, 121 [CrossRef] [Google Scholar]
- Dovesi, R., Erba, A., Orlando, R., et al. 2018, Wiley Interdiscip. Rev. Comput. Mol. Sci., 8, e1360 [CrossRef] [Google Scholar]
- Duley, W. W. 2000, ApJ, 528, 841 [NASA ADS] [CrossRef] [Google Scholar]
- Eckart, C. 1930, Phys. Rev., 35, 1303 [Google Scholar]
- Enrique-Romero, J., & Rimola, A. 2024, https://zenodo.org/records/10518616 [Google Scholar]
- Enrique-Romero, J., Rimola, A., Ceccarelli, C., & Balucani, N. 2016, MNRAS, 459, L6 [Google Scholar]
- Enrique-Romero, J., Rimola, A., Ceccarelli, C., et al. 2019, ACS Earth Space Chem., 3, 2158 [NASA ADS] [CrossRef] [Google Scholar]
- Enrique-Romero, J., Ceccarelli, C., Rimola, A., et al. 2021, A&A, 655, A9 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Enrique-Romero, J., Rimola, A., Ceccarelli, C., et al. 2022, ApJS, 259, 39 [NASA ADS] [CrossRef] [Google Scholar]
- Epping, R. F., Vesseur, D., Zhou, M., & de Bruin, B. 2023, ACS Catal., 13, 5428 [CrossRef] [Google Scholar]
- Ferrante, R. F., Moore, M. H., Nuth, J. A., & Smith, T. 2000, Icarus, 145, 297 [CrossRef] [Google Scholar]
- Ferrero, S., Ceccarelli, C., Ugliengo, P., Sodupe, M., & Rimola, A. 2023a, ApJ, 951, 150 [CrossRef] [Google Scholar]
- Ferrero, S., Pantaleone, S., Ceccarelli, C., et al. 2023b, ApJ, 944, 142 [NASA ADS] [CrossRef] [Google Scholar]
- Fischer, F., & Tropsch, H. 1923, Ber. Dtsch. Chem. Ges., 56, 2428 [CrossRef] [Google Scholar]
- Fischer, F., & Tropsch, H. 1926, Ber. Dtsch. Chem. Ges., 59, 923 [CrossRef] [Google Scholar]
- Foppa, L., Iannuzzi, M., Copéret, C., & Comas-Vives, A. 2018, ACS Catal., 8, 6983 [CrossRef] [Google Scholar]
- Foppa, L., Iannuzzi, M., Copéret, C., & Comas-Vives, A. 2019, ACS Catal., 9, 6571 [CrossRef] [Google Scholar]
- Fredon, A., Groenenboom, G. C., & Cuppen, H. M. 2021, ACS Earth Space Chem., 5, 2032 [NASA ADS] [CrossRef] [Google Scholar]
- Friedel, D. N., & Snyder, L. E. 2008, ApJ, 672, 962 [NASA ADS] [CrossRef] [Google Scholar]
- Fuchs, G. W., Cuppen, H. M., Ioppolo, S., et al. 2009, A&A, 505, 629 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Garrod, R. T., Weaver, S. L. W., & Herbst, E. 2008, ApJ, 682, 283 [NASA ADS] [CrossRef] [Google Scholar]
- Goedecker, S., & Teter, M. 1996, Phys. Rev. B, 54, 1703 [CrossRef] [PubMed] [Google Scholar]
- Grimme, S., Antony, J., Ehrlich, S., & Krieg, H. 2010, J. Chem. Phys., 132, 154104 [Google Scholar]
- Henrici-Olivé, G., & Olivé, S. 1976, Angew. Chem. Int. Ed., 15, 136 [CrossRef] [Google Scholar]
- Herbst, E. 2017, Int. Rev. Phys. Chem., 36, 287 [Google Scholar]
- Herbst, E., & Van Dishoeck, E. F. 2009, ARA&A, 47, 427 [NASA ADS] [CrossRef] [Google Scholar]
- Hollis, J. M., Jewell, P. R., & Lovas, F. J. 1989, ApJ, 346, 794 [NASA ADS] [CrossRef] [Google Scholar]
- Hollis, J. M., Jewell, P. R., & Lovas, F. J. 1995, ApJ, 438, 259 [Google Scholar]
- Huff, G. A., & Satterfield, C. N. 1984, Ind. Eng. Chem. Process. Des. Dev., 23, 696 [CrossRef] [Google Scholar]
- Inostroza-Pino, N., Macleod-Carey, D., Heyser, C., et al. 2021, A&A, 650, A169 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jin, M., & Garrod, R. T. 2020, ApJS, 249, 26 [Google Scholar]
- Jørgensen, J. K., Favre, C., Bisschop, S. E., et al. 2012, ApJ, 757, L4 [Google Scholar]
- Keller, L. P., Hony, S., Bradley, J. P., et al. 2002, Nature, 417, 148 [Google Scholar]
- Khodakov, A. Y., Chu, W., & Fongarland, P. 2007, Chem. Rev., 107, 1692 [CrossRef] [Google Scholar]
- Kühne, T. D., Iannuzzi, M., Del Ben, M., et al. 2020, J. Chem. Phys., 152 [Google Scholar]
- Kummer, J. T., & Emmett, P. H. 1953, J. Am. Chem. Soc., 75, 5177 [CrossRef] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Li, H. J., Chang, C. C., & Ho, J. J. 2011, J. Phys. Chem. C, 115, 11045 [NASA ADS] [CrossRef] [Google Scholar]
- Lippert, G., Hutter, J., & Parrinello, M. 1997, Mol. Phys., 92, 477 [NASA ADS] [CrossRef] [Google Scholar]
- Llorca, J., & Casanova, I. 1998, Meteor. Planet. Sci., 33, 243 [NASA ADS] [CrossRef] [Google Scholar]
- Lodders, K., & Fegley, B. 2023, Geochemistry, 83, 1220 [Google Scholar]
- Marchione, D., Rosu-Finsen, A., Taj, S., et al. 2019, ACS Earth Space Chem., 3, 1915 [NASA ADS] [CrossRef] [Google Scholar]
- Marcus, R. A. 1952, J. Chem. Phys., 20, 359 [NASA ADS] [CrossRef] [Google Scholar]
- Masters, C. 1979, Adv. Organomet. Chem., 17, 61 [CrossRef] [Google Scholar]
- McClure, M. K., Rocha, W. R. M., Pontoppidan, K. M., et al. 2023, Nat. Astron., 7, 431 [NASA ADS] [CrossRef] [Google Scholar]
- Millar, T. J., MacDonald, G. H., & Habing, R. J. 1995, MNRAS, 273, 25 [NASA ADS] [CrossRef] [Google Scholar]
- Molpeceres, G., Zaverkin, V., Furuya, K., Aikawa, Y., & Kästner, J. 2023, A&A, 673, A51 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Nuth III, J. A., Charnley, S. B., & Johnson, N. M. 2006, in Meteorites and the Early Solar System II (University of Arizona Press), 147 [CrossRef] [Google Scholar]
- Öberg, K. I., Boogert, A. C., Pontoppidan, K. M., et al. 2011, ApJ, 740, 109 [CrossRef] [Google Scholar]
- Öberg, K. I., Facchini, S., & Anderson, D. E. 2023, ARA&A, 61, 287 [CrossRef] [Google Scholar]
- Ohishi, M., Ishikawa, S.-I., Yamamoto, S., Saito, S., & Amano, T. 1995, ApJ, 446, L43 [NASA ADS] [CrossRef] [Google Scholar]
- Palau, A., Fuente, A., Girart, J. M., et al. 2011, ApJ, 743, L32 [NASA ADS] [CrossRef] [Google Scholar]
- Pantaleone, S., Enrique-Romero, J., Ceccarelli, C., et al. 2021, ApJ, 917, 49 [CrossRef] [Google Scholar]
- Pareras, G., Cabedo, V., McCoustra, M., & Rimola, A. 2023, A&A, 680, A57 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Perdew, J. P., Ruzsinszky, A., Csonka, G. I., et al. 2008, Phys. Rev. Lett., 100, 136406 [CrossRef] [PubMed] [Google Scholar]
- Perrero, J., Enrique-Romero, J., Martínez-Bachs, B., et al. 2022, ACS Earth Space Chem., 6, 496 [NASA ADS] [CrossRef] [Google Scholar]
- Peters, S., Semenov, D. A., Hochleitner, R., & Trapp, O. 2023, Sci. Rep., 13, 1 [CrossRef] [Google Scholar]
- Pizzarello, S. 2006, Acc. Chem. Res., 39, 231 [CrossRef] [Google Scholar]
- Potapov, A., Jäger, C., Henning, T., Jonusas, M., & Krim, L. 2017, ApJ, 846, 131 [Google Scholar]
- Potapov, A., Jäger, C., & Henning, T. 2020, Phys. Rev. Lett., 124, 221103 [Google Scholar]
- Psarras, P. C., & Ball, D. W. 2015, Comput. Theor. Chem., 1063, 1 [CrossRef] [Google Scholar]
- Rittmeyer, S. P., Bukas, V. J., & Reuter, K. 2018, Adv. Phys. X, 3, 1381574 [NASA ADS] [Google Scholar]
- Rivilla, V. M., Beltrán, M. T., Martín-Pintado, J., et al. 2017, A&A, 599, A26 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Roberts, H., Herbst, E., & Millar, T. J. 2004, A&A, 424, 905 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rocha, W. R. M., van Dishoeck, E. F., Ressler, M. E., et al. 2024, A&A, 683, A124 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rubin, R. H., Swenson, G. W., J., Benson, R. C., Tigelaar, H. L., & Flygare, W. H. 1971, ApJ, 169, L39 [Google Scholar]
- Saladino, R., Botta, G., Pino, S., Costanzo, G., & Di Mauro, E. 2012, Chem. Soc. Rev., 41, 5526 [CrossRef] [Google Scholar]
- Sandford, S. A., Aléon, J., Alexander, C. M., et al. 2006, Science, 314, 1720 [NASA ADS] [CrossRef] [Google Scholar]
- Santamaría, J., & Aguilar, E. 2016, Org. Chem. Front., 3, 1561 [CrossRef] [Google Scholar]
- Sargent, B. A., Forrest, W. J., Tayrien, C., et al. 2009, ApJ, 690, 1193 [NASA ADS] [CrossRef] [Google Scholar]
- Sasaki, S., Nakamura, K., Hamabe, Y., Kurahashi, E., & Hiroi, T. 2001, Nature, 410, 555 [NASA ADS] [CrossRef] [Google Scholar]
- Schulz, H. 1999, Appl. Catal. A-GEN., 186, 3 [CrossRef] [Google Scholar]
- Sekine, Y., Sugita, S., Shido, T., et al. 2006, Meteor. Planet. Sci., 41, 715 [NASA ADS] [CrossRef] [Google Scholar]
- Skouteris, D., Balucani, N., Ceccarelli, C., et al. 2018, ApJ, 854, 135 [NASA ADS] [CrossRef] [Google Scholar]
- Stephens, P. J., Devlin, F. J., Chabalowski, C. F., & Frisch, M. J. 1994, J. Phys. Chem., 98, 11623 [CrossRef] [Google Scholar]
- Taquet, V., López-Sepulcre, A., Ceccarelli, C., et al. 2015, ApJ, 804, 81 [Google Scholar]
- Ugliengo, P., Sodupe, M., Musso, F., et al. 2008, Adv. Mater., 20, 4579 [NASA ADS] [CrossRef] [Google Scholar]
- Van Der Laan, G. P., & Beenackers, A. A. 1999, Catal. Rev. Sci. Eng., 41, 255 [CrossRef] [Google Scholar]
- Vannice, M. A. 1976, Catal. Rev. Sci. Eng., 14, 153 [CrossRef] [Google Scholar]
- Vasyunin, A. I., & Herbst, E. 2013, ApJ, 769, 34 [Google Scholar]
- Vasyunin, A. I., Caselli, P., Dulieu, F., & Jiménez-Serra, I. 2017, ApJ, 842, 33 [Google Scholar]
- Vazart, F., Ceccarelli, C., Balucani, N., Bianchi, E., & Skouteris, D. 2020, MNRAS, 499, 5547 [Google Scholar]
- Walsh, C., Millar, T. J., Nomura, H., et al. 2014, A&A, 563, A33 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Watanabe, N., & Kouchi, A. 2002, ApJ, 571, L173 [Google Scholar]
- Zhuo, M., Tan, K. F., Borgna, A., & Saeys, M. 2009, J. Phys. Chem. C, 113, 8357 [NASA ADS] [CrossRef] [Google Scholar]
- Zuckerman, B., Turner, B. E., Johnson, D. R., et al. 1975, ApJ, 196, L99 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
All Figures
|
Fig. 1 Schematic representation of the FTT processes simulated in this work. Green line: C-O dissociation of acetaldehyde (CH2O), forming the CH2 chain initiator and O, and H2 addition to atomic O to form H2O. Blue lines: formation of ethanol (CH3CH2OH) by (i) reaction of CH2 with CO, forming ketene (CH2CO), and (ii) two H2 additions to ketene to form first acetaldehyde (CH3CHO) and finally ethanol. Red lines: formation of methane (CH4) by H2 addition to CH2. |
| In the text | |
|
Fig. 2 Top: scheme of the Fe0 grafting on the SiO2 surface. Bottom: zoom-out (left) and zoom-in (right) of the optimized geometry of the Fe0 @SiO2 system. The dashed line box represents the atoms included in the partial Hessian matrix calculation. Relevant distances are depicted in Angstroms (Å). Color-coding: H atoms are represented in white, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
| In the text | |
|
Fig. 3 (A) Reaction mechanism for the CO dissociation into C + O. (B) Reaction mechanism for the HCO dissociation into HC + O. (C) Reaction mechanism for the H2CO dissociation into H2C and O. (D) Reaction mechanism for the first oxygen hydrogenation to OH. (E) Reaction mechanism for the second oxygen hydrogenation to H2O. (F) ZPE-corrected PESs (in kcal mol–1) for the different proposed CO dissociation mechanisms, as well as of H2O and CH3OH formation, using as the 0th reference energy the Fe0@SiO2 + H2 + CO asymptote. The optimized geometries of the five transition state structures involved in the three different C-O dissociations and in the H2O formation are also shown (bond distances in Å). The asterisk symbol (*) denotes from which atom the relevant molecule is coordinated with Fe. Color-coding: H atoms are represented in white, C atoms in gray, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
| In the text | |
|
Fig. 4 (A) Reaction mechanism for the ethanol formation from the direct CH2 – CO coupling forming CH2 and O, the reaction of CH2 with an incoming CO, forming H2CCO, and subsequent hydrogenations. (B) Reaction mechanism for the methane formation from hydrogenation of CH2. (C) ZPE-corrected PESs (in kcal mol–1 ) for the ethanol formation (black) and methane formation (gray) mechanisms, using as the 0th reference energy the Fe0@SiO2-CH2 + H2 + CO asymptote. The optimized geometries of all the transition state structures (bond distances in Å) are also shown. The asterisk symbol (*) denotes from which atom the relevant molecule is coordinated with Fe. Color-coding: H atoms are represented in white, C atoms in gray, O atoms in red, Si atoms in beige, and Fe atoms in orange. |
| In the text | |
|
Fig. 5 Arrhenius plots for the elementary steps associated with |
| In the text | |
|
Fig. A.1 Representation of the optimized unit cell considered in this work. |
| In the text | |
|
Fig. D.1 Representation of the calculated frontier orbitals, from left to right: SOMO-1, SOMO, and LUMO. |
| In the text | |
|
Fig. E.1 Arrhenius plot for the free energy barrier associated with TSC–O (panel A), TSHC–O (panel B), TSH2C–O (panel C), TSCH2OH (panel D), and TSH2C–OH (panel E). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
| In the text | |
|
Fig. E.2 Arrhenius plot for the free energy barrier associated with TSO–H (panel A) and TSHO–H (panel B). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
| In the text | |
|
Fig. E.3 Arrhenius plot for the free energy barrier associated with TS1 (panel A), TS2 (panel B), TS3 (panel C), TS4 (panel D), and TS5 (panel E). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1. Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
| In the text | |
|
Fig. E.4 Arrhenius plot for the free energy barrier associated with TS6 (panel A) and TS7 (panel B). Values of the natural logarithm of the rate constant (ln(k)) are represented in year–1 and the inverse of the temperatures (1/T) in K–1 . Represented in blue are the RRKM rate constants and in orange the rate constants, considering tunneling. Also included are the fits and correlation coefficients. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.