| Issue |
A&A
Volume 689, September 2024
|
|
|---|---|---|
| Article Number | A21 | |
| Number of page(s) | 14 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202450649 | |
| Published online | 28 August 2024 | |
H-atom-assisted formation of key radical intermediates in interstellar sugar synthesis
New insights from para-H2 matrix isolation experiments
1
Laboratory of Molecular Spectroscopy, Institute of Chemistry, ELTE Eötvös Loránd University,
PO Box 32,
1518
Budapest,
Hungary
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2
Hevesy György PhD School of Chemistry, Institute of Chemistry, ELTE Eötvös Loránd University,
PO Box 32,
1518
Budapest,
Hungary
3
Laboratory of Theoretical Chemistry, Institute of Chemistry, ELTE Eötvös Loránd University,
1117,
Budapest,
Hungary
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
4
MTA-ELTE Lendület Laboratory Astrochemistry Research Group, Institute of Chemistry, ELTE Eötvös Loránd University,
PO Box 32,
1518
Budapest,
Hungary
5
Present address: School of Advanced Sciences (SAS), Vellore Institute of Technology, Vellore Campus, Vellore
632014,
Tamil Nadu,
India
6
Centre for Astrophysics and Space Science, ELTE Eötvös Loránd University,
PO Box 32,
1518
Budapest,
Hungary
Received:
8
May
2024
Accepted:
18
June
2024
Abstract
Context. Despite the identification of the smallest sugar molecule, glycolaldehyde (GA), in the interstellar medium (ISM), its mechanism of formation in the ISM is still not fully understood. A more profound understanding of the interstellar chemistry of GA and related molecules could provide insights into whether larger sugar molecules can also form and survive under such conditions.
Aims. The primary objectives of this research are to delve into the sugar formation mechanism in the ISM, especially in dark molecular clouds; unravel intricate details of H-atom-mediated reactions involving glyoxal (GO), GA, and ethylene glycol (EG); and identify intermediates playing potential roles in the formation of larger sugars or serving as intermediates in the destruction reaction paths of sugar molecules.
Methods. The study utilizes the para-H2 matrix isolation method with infrared (IR) spectroscopic detection and quantum chemical computations to investigate H-atom reactions of GO, GA, and EG at a low temperature.
Results. Several radical products were spectroscopically identified that might be key active species in the interstellar formation of larger sugar molecules.
Key words: astrochemistry / dense matter / molecular processes / ISM: molecules
© The Authors 2024
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
Carbohydrates or sugars are essential chemical substances for Earth-type life. For several decades, scientists have explored the fascinating question of how sugars formed upon chemical evolution from smaller molecules, unraveling the transformation of simple building blocks. In the realm of carbohydrate formation, the formose reaction (FR) holds a significant historical position as the first discovered route of monosaccharide synthesis. Initiated by the pioneering work of A. M. Butlerov in 1861, the FR involves the oligomerization of formaldehyde in aqueous solution in the presence of basic catalysts, accompanied by numerous disproportionation and cleavage reactions (Butlerow 1861). The classical FR yields a diverse mixture of different sugars. Years of intensive research have yielded significant advancements in comprehending the kinetics, mechanisms, selectivity, and catalytic reaction pathways associated with the FR, providing insights into conditions resembling those of early Earth (Khomenko et al. 1980; Mizuno & Weiss 1974; Orgel 1998).
Since the FR requires liquid water, it naturally prompts intriguing questions about the presence and formation of small carbohydrates in cold environments of the Universe in which liquid water is absent. Glycolaldehyde (GA, O=CH−CH2−OH), often considered to be the simplest carbohydrate (Aviles-Moreno et al. 2006; Fedoseev et al. 2017; Jørgensen et al. 2012; Li et al. 2022), has been detected in various astrophysical environments. GA was first observed in Sgr B2(N), in which the column density of both formaldehyde (H2C=O) and methanol (CH3−OH) is high (Houghton & Whiteoak 1995; Hollis et al. 2000; Halfen et al. 2006; Hoffman et al. 2007). Subsequently, GA has been observed in diverse systems, including the hot molecular core G31.41 + 0.31 (Beltrán et al. 2009), the Class 0 protostellar binary IRAS 16293-2422 (Jørgensen et al. 2012), and the NGC 1333-IRAS2A solar-type protostar (Coutens et al. 2015). GA has been identified in multiple sources of the Perseus molecular cloud (De Simone et al. 2017) and in the shock region L1157-B1 (Lefloch et al. 2017). It has also been detected in comets like Lovejoy (Biver et al. 2015) and 67P/Churyumov–Gerasimenko (Goesmann et al. 2015). Additionally, the saturated – that is, hydrogenated – form of GA, ethylene glycol (EG, HO−CH2−CH2−OH), has been detected toward the Galactic center (Hollis et al. 2002) and later in NGC 1333-IRAS2A protostar (Maury et al. 2014). It is considered one of the largest molecules identified in a comet, notably in the comet C/1995 O1 (Hale-Bopp) (Crovisier et al. 2004). Therefore, the astrophysical observations clearly indicate that liquid water-free mechanisms for carbohydrate formation must exist in cold astrophysical environments (Woods et al. 2012).
The first gas-phase mechanism proposed for the interstellar formation of GA included four steps: the protonation of H2C=O by ![Mathematical equation: $\[\mathrm{H}_3^{+}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq1.png) , the reaction of H2C−OH+ by another H2C=O, a rearrangement step, and finally deprotonation (Halfen et al. 2006). Due to the considerable reaction barriers in this GA formation mechanism, this route is not efficient (Woods et al. 2012, 2013), which prompted the exploration of alternative mechanisms. According to one of these new models, hydroxycarbene (
, the reaction of H2C−OH+ by another H2C=O, a rearrangement step, and finally deprotonation (Halfen et al. 2006). Due to the considerable reaction barriers in this GA formation mechanism, this route is not efficient (Woods et al. 2012, 2013), which prompted the exploration of alternative mechanisms. According to one of these new models, hydroxycarbene (![Mathematical equation: $\[\mathrm{H} \ddot{\mathrm{C}}{-}\mathrm{OH}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq2.png) ), generated through the photolysis of carbon monoxide (CO) and a hydrogen molecule (H2), can react in a nearly barrierless reaction with formaldehyde (H2C=O), producing GA (Eckhardt et al. 2019; Mardyukov et al. 2023). Recent theoretical computations suggest that GA can be formed at a nonzero rate by a quantum tunneling reaction from two H2C=O (Ahmad et al. 2020). According to another recent gas-phase model, GA (Skouteris et al. 2018) can be formed from ethanol (CH3−CH2−OH) in a two-step reaction sequence. First, a radical abstracts an H atom from CH3−CH2−OH, generating the 2-hydroxyethyl (ĊH2−CH2−OH) radical. Then, the reaction of an O atom with ĊH2−CH2−OH produces GA and an H atom. While these mechanisms can potentially enhance the efficiency of GA formation, some have not been tested in simulations, and for those that have, the simulations show that the mechanisms are still insufficient to explain the observed column densities of GA; for example, in hot cores (Silva et al. 2020). More importantly, astronomical observations revealed a significant correlation between the column densities of GA and its isomers, acetic acid (CH3−C(O)−OH) and methyl formate (O=CH−O−CH3), as well as EG. Thus, these species are chemically related, which cannot be explained by the existing gas-phase models. Consequently, these findings redirected research toward ice grain or grain surface-gas models.
), generated through the photolysis of carbon monoxide (CO) and a hydrogen molecule (H2), can react in a nearly barrierless reaction with formaldehyde (H2C=O), producing GA (Eckhardt et al. 2019; Mardyukov et al. 2023). Recent theoretical computations suggest that GA can be formed at a nonzero rate by a quantum tunneling reaction from two H2C=O (Ahmad et al. 2020). According to another recent gas-phase model, GA (Skouteris et al. 2018) can be formed from ethanol (CH3−CH2−OH) in a two-step reaction sequence. First, a radical abstracts an H atom from CH3−CH2−OH, generating the 2-hydroxyethyl (ĊH2−CH2−OH) radical. Then, the reaction of an O atom with ĊH2−CH2−OH produces GA and an H atom. While these mechanisms can potentially enhance the efficiency of GA formation, some have not been tested in simulations, and for those that have, the simulations show that the mechanisms are still insufficient to explain the observed column densities of GA; for example, in hot cores (Silva et al. 2020). More importantly, astronomical observations revealed a significant correlation between the column densities of GA and its isomers, acetic acid (CH3−C(O)−OH) and methyl formate (O=CH−O−CH3), as well as EG. Thus, these species are chemically related, which cannot be explained by the existing gas-phase models. Consequently, these findings redirected research toward ice grain or grain surface-gas models.
A grain surface mechanism was formulated by Sorrell (2001). According to this, the formyl radical (HĊ=O) formed by the reaction of an H atom and CO, can react with methanol (CH3−OH), producing GA and an H atom. Beltrán et al. (2009) suggested a similar surface mechanism, the reaction of hydroxy methyl radical (ĊH2−OH), HĊ=O, and H. Bennett & Kaiser (2007) and Maity et al. (2015), mimicking the effect of cosmic rays, conducted electron irradiation experiments on methanol (CH3−OH):CO ices. They observed the formation of both GA and O=CH−O−CH3. According to their model, methoxy (CH3−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq3.png) ) and hydroxy methyl (ĊH2−OH) radicals together with H atoms are formed in the first step. The H atoms and CO, which form HĊ=O and the recombination of HĊ=O with CH3−
) and hydroxy methyl (ĊH2−OH) radicals together with H atoms are formed in the first step. The H atoms and CO, which form HĊ=O and the recombination of HĊ=O with CH3−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq4.png) or ĊH2−OH, lead to the formation of O=CH−O−CH3 and GA, respectively. Therefore, this model explains not only the formation of GA but also its chemical link with O=CH−O−CH3.
or ĊH2−OH, lead to the formation of O=CH−O−CH3 and GA, respectively. Therefore, this model explains not only the formation of GA but also its chemical link with O=CH−O−CH3.
The radical recombination mechanism involving HĊ=O and ĊH2−OH radicals has been extended by several studies. An important realization was that while the H + CO reaction is not sufficiently exothermic for HĊ=O to desorb from the surface (Pantaleone et al. 2020), and thus continue reacting with the ĊH2−OH radical, the second reaction is exothermic enough to transfer the final GA product to the gaseous phase of the interstellar medium (ISM) (Paiva et al. 2023). Woods et al. (2013) proposed an alternative formation route for GA, the recombination of two HĊ=O radicals, yielding glyoxal (GO, O=CH−CH=O), followed by two H-atom additions. Fedoseev et al. (2015), Chuang et al. (2016), and Coutens et al. (2018) expanded on this model by introducing the possibility of two ĊH2−OH radicals recombining to form EG. This model states that GO, GA, and EG are interconnected through successive H atom additions. Subsequent laboratory studies further confirmed the general picture but also identified some problems with this model. While experiments involving the preparation of HĊ=O and ĊH2−CH radicals in an inert Ar matrix, followed by heating, confirmed the recombination of HĊ=O and ĊH2−OH to form GA (Butscher et al. 2015), analogous experiments with only HĊ=O radicals failed to produce GO (Butscher et al. 2017). Quantum chemical computations unveiled that once GO is formed by radical-radical recombination, its exothermic nature induces intramolecular H atom transfer, resulting in the formation of H2C=O and CO. Additionally, intermolecular H atom transfer between two HĊ=O species also produces the same products (Butscher et al. 2017). Further experiments indicated the complexity of the H atom addition reaction sequence from GO to EG. While H atoms reacting with GA ice at 10 K led to the formation of EG (Leroux et al. 2021), H atoms in GO ice produced CO and H2C=O instead of H-addition products like GA (Leroux et al. 2020). The latter observation was explained by subsequent H addition to one of the C atoms and H atom abstraction from the other C atom, followed by C−C bond rupture. These observations strongly suggest that the formation of GO is decoupled from the formation network of GA and EG, which is consistent with the non-observation of GO in the ISM.
The radical-radical recombination model has been extended to larger sugar molecules and experimentally verified by Fedoseev et al. (2017) in cold prestellar cores, in which H atoms are present but the penetration of UV radiation is limited. In their experiment, CO, GA, and H atoms were co-deposited, leading to the formation of glycerol (HO−CH2−CH2(OH)−CH2−OH) and, tentatively, glyceraldehyde (HO−CH2−CH2(OH)−CH=O) in the ice. Subsequent experiments irradiating an H2C=O:H2O mixture with UV photons have revealed that both of these products can form in environments in which radiation drives the chemical processes (Layssac et al. 2020). Although reactive intermediates could not be spectroscopically observed in the processed ices, plausible mechanisms were suggested based on the analogy of EG and GA formation. Fedoseev et al. (2015) proposed the formation of the HO−CH2−ĊH−OH radical, recombining with HĊ=O or ĊH2−OH radicals, resulting in the formation of HO−CH2−CH2(OH)−CH=O or HO−CH2−CH2(OH)−CH2−OH, respectively. In the latter study, the reaction of the HO−ĊH−CH=O radical with CH2=O was proposed, with subsequent hydrogenation steps leading to HO−CH2−CH(OH)−CH=O or HO−CH2−CH(OH)−CH2−OH (Layssac et al. 2020).
In a recent laboratory study, the formation of the enolic form of GA, 1,2-ethenediol (HO−CH=CH−OH) was observed upon the electron irradiation of CH3−OH:CO ice (Kleimeier et al. 2021). It was proposed that this species could be the active form in the FR. Furthermore, the computed 24 kJ mol−1 barrier of the reaction between 1,2-ethenediol and GA to form glyceraldehyde might be low enough to occur even at the low temperatures found in the ISM, either through tunneling or energetic cosmic ray processing. Subsequently, 1,2-ethenediol was identified by microwave spectroscopy in the laboratory (Melosso et al. 2022), and then by radioastronomical observation in the ISM toward the G+0.693−0.027 molecular cloud (Rivilla et al. 2022).
The objective of this study is to delve deeper into the low-temperature chemistry mediated by H atoms involving GO, GA, and EG. Differing from previous investigations, we employed the para-H2 matrix-isolation method in this work. Despite the apolar and colder characteristics of the para-H2 matrix compared to typical interstellar ice environments, it offers notable advantages. This matrix facilitates the generation and movement of H atoms through quantum diffusion, thereby enabling effective reactions with the molecules under investigation. The infrared (IR) spectra exhibit sharp bands, providing exceptional resolution and clear assignment – an aspect that can be challenging in analogous astrophysical ices. These advantages enable the identification of reactive intermediates that might not be conclusively discernible in astrophysical analog ices. Such intermediates include those connecting GO, GA, and EG chemically, those through which H atoms induce the decomposition of these species, or intermediates that might play an active role in the interstellar formation mechanism of sugars. In summary, through the application of the para-H2 matrix-isolation method combined with quantum-chemical computations, this study aims to contribute to our comprehension of the chemical mechanisms involved in sugar formation within the ISM, focusing on the H-atom-induced chemical reactions of GO, GA, and EG.
2 Materials and methods
All experiments were performed using the ultra-high vacuum (UHV) compatible VIZSLA experimental setup (with a base pressure of ≈10−8 mbar when not cooled) (Bazsó et al. 2021). The experiments were done in a para-H2 matrix; for this, the ortho-H2−para-H2 conversion had to be executed by letting normal−H2 (Messer, 99.999%) flow through a capillary filled with an Fe2O3 catalyst (SigmaAldrich, hydrated, catalyst grade, 30–50 mesh). The capillary was wrapped around the cold head of the “upper” cryostat (CH202, Sumitomo Heavy Industries Inc.) of the setup and was cooled to T = 13.9 K, which ensured a rapid and efficient ortho-to-para conversion rate. The para-H2 was collected in a 0.5 L glass flask on a vacuum line coupled to the VIZSLA setup.
The samples as well as para-H2 were co-deposited on a gold-coated silver wafer serving as substrate in the main chamber of the experimental setup. The substrate is mounted on the cold head of the “lower” cryostat (RDK-415D, Sumitomo Heavy Industries Inc.) of the setup and was eventually cooled down to T = 3.1 K. In order to obtain the vapor pressure needed for the desired nominal sample – a para-H2 mixing ratio of 1:2000 – the samples had to be kept at different temperatures. To produce monomeric GO, its trimer dihydrate precursor (Santa Cruz Biotechnology, ≥95%) was heated up to 102–107 °C. The sample temperature was kept at 36–38 °C for GA dimer (Merck, mixture of stereoisomers) to produce vapors containing the monomeric form. The samples were placed into a small glass vial directly attached to the main chamber of the setup and were heated by a hot plate using water or a silicon oil bath depending on the desired sample temperature. As for EG (Merck, ReagentPlus, ≥99%), it had to be cooled to 0–5 °C using a water bath. In every case, the sample temperatures were measured by a thermocouple. The sample vapors enter the main chamber through a stainless steel feed-trough ending in a teflon tube 6 mm in diameter; the distance between the tip and the substrate is ca 3 cm. The para-H2 inlet occurred through a leak valve and a stainless steel capillary array also ending some 3 cm before the substrate, allowing for its proper mixing with the sample before deposition. Molecular Cl2 also had to be introduced using another leak valve and capillary array for the H-atom-generation process. The Cl2 : para-H2 ratio was approximately 1:700–1:800 in each experiment.
The H atoms were generated following the two-step process (Raston & Anderson 2006, 2007; Kettwich et al. 2009; Raston et al. 2010, 2015), which was successfully implemented in our laboratory (Bazsó et al. 2021; Góbi et al. 2021; Schneiker et al. 2021, 2022; Keresztes et al. 2023). For this, the deposited matrices were subjected to 365 nm LED irradiation (M365L3 LED source, ThorLabs, fwhm ≃ 9 nm, equipped with an SM1U collimation adapter and a LEDD1B driver) for 60–90 min, applying a current of I = 0.16–0.50 A to make the Cl2 molecules dissociate, and thus generating Cl atoms. It is worth mentioning that owing to the greatly diminished cage effect of the para-H2 matrix, (Tsuge & Lee 2020), the two formed Cl atoms quickly separate and cannot recombine. Afterwards, by the 2217 nm (near-infrared; NIR) laser excitation of the Q1(1) + S0(0) and Q1(0) + S0(0) combination bands of the para-H2 molecules makes the Cl + H2 viable yielding H atoms and HCl. Then, the matrix was kept in the dark overnight, which is an important phase as the H atoms present after the irradiation had sufficient time to diffuse in the soft matrix and participate in hydrogenation reactions. The hydrogenation process was repeated the following day. Lastly, the matrices underwent a set of secondary laser UV photolyses. These experiments were conducted to study the photophysical behavior of the sample molecules and the radicals formed during the tests. Additionally, the series of UV irradiations supported the assignment of the vibrational bands based on their responses to different UV wavelengths, as bands corresponding to the same molecule should exhibit the same behavior when exposed to the same wavelength of irradiation. The utilized laser wavelengths were 300, 270, 240, and 216 nm, respectively, each lasting roughly 30 min. The different laser irradiations were performed using an optical parametric oscillator (OPO, GWU / Spectra-Physics VersaScan MB 240, fwhm ≃ 5 cm−1) equipped with a frequency-doubling unit (Spectra-Physics uvScan) pumped by a pulsed Nd:YAG laser (Spectra-Physics Quanta Ray Lab 150, P ≈ 2.1–2.2 W, λ = 355 nm, f = 10 Hz, pulse duration = 2–3 ns).
The changes occurring during the experiments were detected by the help of a Bruker Invenio Fourier-transform infrared (FTIR) spectrometer working in reflection-absorption mode and using a liquid-N2-cooled mercury cadmium telluride (MCT) detector. The spectral resolution was set to 0.5 cm−1 and the spectra were collected in the 4000–600 cm−1 spectral range, whereas the NIR spectra were taken in the 9000–600 cm−1 spectral ranges. To follow the sample deposition, MIR spectra were collected each minute, averaging 32 scans. Subsequently, a 32-scan NIR spectrum was taken to check the purity of the para-H2 matrix through the 4740 cm−1 band of the ortho-isomer, which can be attributed to its Q1(0) + S0(1) transition. The spectrometer beam was diverted during the 365 nm irradiation to prevent NIR excitation induced by the spectrometer beam. Every 30 min, the LED irradiation was stopped to monitor the Cl atom formation based on the growth of the 5095 cm−1 band by averaging 32 scans. The formation of the H atoms during the 2217 nm laser irradiation could be followed only indirectly, by assessing the vibrational absorption peak of the other product (HCl) at 2894 cm−1. For this, 32 scans were saved each minute in the MIR region using a long-pass filter with a cutoff wavelength of 3860 cm−1 to avoid the interference of the NIR laser photons with the MCT detector. 32-scan MIR spectra were taken during the secondary UV irradiation studies as well with the same cutoff filter in the spectrometer beam path. Longer MIR spectra were collected between the different phases of the experiment, averaging 128 scans. Blank experiments were also run when no Cl2 molecules were present in the matrix. Otherwise, the experiment was similar to the real ones, except that both the 365 nm LED and the 2217 nm laser NIR irradiation took only 30 min. The blank experiments allowed us to evaluate the photolytic effect of the irradiation, especially the 365 nm one on the matrix.
To predict the reaction paths and aid spectral assignments quantum chemical computations were carried out. First, the minimum-energy structures and the transition states were located on the potential energy surface (PES) at the B3LYP/cc-pVTZ level of theory. Then, at the same level, harmonic and anharmonic vibrational frequencies and IR intensities were computed by vibrational perturbation theory (VPT2); these are listed in the supporting information in Tables S1–S54. To ensure that the transition structures connected the right minima, intrinsic reaction coordinate (IRC) calculations were performed. To get better estimates of vibrational frequencies, geometry optimizations were also performed at the frozen-core (core-electrons were not correlated) CCSD(T)/cc-pVTZ level of theory, followed by harmonic vibrational frequency computations (see the supporting information in Tables S55–S62 for the details of the experimentally identified species, detailed in Sec. 3). The best estimate for the vibrational frequency of each normal mode was obtained as the sum of CCSD(T)/cc-pVTZ harmonic vibrational frequency and B3LYP/cc-pVTZ anharmonic correction. The electronic structure computations at the B3LYP/cc-pVTZ and the CCSD(T)/cc-pVTZ levels were executed with Gaussian 09 (rev. D01) (Frisch et al. 2009) and CFOUR (Matthews et al. 2020) program packages, respectively.
Álvarez-Barcia et al. (2018) theoretically investigated the H-atom reactions of GO, GA, and EG, employing advanced computational methods (MPWB1K/def2-TZVP level of theory). However, their analysis had no regard for various conformers. Thus, we provide a comprehensive summary of computational details encompassing the reactions of the different conformers. In the following, the prefixes trans and cis in the names of the different species refer to the position of the ligands around the C−C bond (as it is used in the literature), while the prefixes anti and syn are used to characterize the CCOH torsion in the molecules.
3 Results and discussion
3.1 Glyoxal (GO)
Glyoxal exists in two isomeric forms; namely, trans-GO and cis-GO (Bock et al. 1988). The trans-GO configuration exhibits greater stability, surpassing cis-GO by 18.3 kJ mol−1 (see Fig. 1). HO−C≡C−OH can be considered a keto-enol tautomer of GO, which has energy 188.7 kJ mol−1 higher than trans-GO. Another tautomeric form of GO is hydroxyketene (HK, O=C=CH−OH). According to theory, HK is more stable than trans-GO by 42.1 kJ mol−1. HK could be formed through an intramolecular tautomerization; however, the tautomerisation reaction has a high barrier of 238.3 kJ mol−1. Consistent with these computational values, and in agreement with previous experiments conducted in an Ar matrix (Engdahl & Nelander 1988), upon evaporation of GO and deposition into para-H2 matrix, only trans-GO was observed. Its prominent IR absorption bands manifest at 2841.8, 1728.9, and 1313.9 cm−1. The full list of the vibrational transitions observed in para-H2 together with the Ar matrix values (Engdahl & Nelander 1988) are listed in Table S63 of the supporting information.
Before analyzing the experiments on the GO + H reactions, we briefly discuss the theoretical predictions of the possible reaction paths, which are shown in Fig. 2. Given that only one isomer, specifically trans-GO, was present in the deposited para-H2 matrix during the experiments, we focus here solely on the reactions involving trans-GO. Computational data for cis-GO can be found in the supporting information (Table S64).
Firstly, trans-GO can engage in a barrierless H-atomabstraction reaction, determined at the B3LYP/cc-pVTZ level of theory, resulting in the formation of the O=Ċ−CH=O radical (R1). There are two possible H-atom-addition reactions of trans-GO: the reaction can proceed either at the O atom or the C atom of the molecule. The computed barriers to the formation of syn,trans−HO−ĊH−CH=O (syn,trans-R2), anti,trans−HO−ĊH−CH=O (anti,trans-R2), and trans−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq5.png) −CH2−CH=O (trans-R3) are 16.5, 16.5, and 10.7 kJ mol−1, respectively. The energy diagram illustrating these potential GO + H reactions is depicted in Fig. 3, and the energies shown in Fig. 3 are listed in Table 1. The computations conducted by Álvarez-Barcia et al. (2018) yielded results consistent with our outlined calculations.
−CH2−CH=O (trans-R3) are 16.5, 16.5, and 10.7 kJ mol−1, respectively. The energy diagram illustrating these potential GO + H reactions is depicted in Fig. 3, and the energies shown in Fig. 3 are listed in Table 1. The computations conducted by Álvarez-Barcia et al. (2018) yielded results consistent with our outlined calculations.
Given the large-quantity generation of H-atoms (845 ppm) in our experiments, a second H-atom reaction becomes plausible, which can be either an H-atom-abstraction or an H-atom-addition reaction. Computations indicate that all second H-atom reactions are barrierless. The comprehensive scheme of potential reactions is illustrated in Fig. 2. Focusing on reactions connecting GO and GA, it is noteworthy that two crucial species – namely, R2 and R3 – can serve as the link in the formation between these two molecules. In addition, other important species, hydroxyketene (HK, O=C=CH−OH), 1,2-ethenediol (ED, OH−CH=CH−OH), and ![Mathematical equation: $\[\mathrm{HO}{-}\ddot{\mathrm{C}}{-}\mathrm{CH}{=}\mathrm{O}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq6.png) (CB1), can serve as a possible indirect connection link between GO and GA.
(CB1), can serve as a possible indirect connection link between GO and GA.
After the deposition, H atoms were generated in the matrix, and the reaction between GO and H was investigated. The generation of H atoms was carried out in a two-step process, described in Sect. 2, with the first step involving 365 nm UV irradiation. The amounts of CO and HĊ=O started to increase in the matrix upon this UV irradiation. CO was identified according to its vibrational band at 2143.2 cm−1, while HĊ=O was identified at 1865.0 cm−1 (see Tables S65 and S66 in the supporting information). The observed vibrational frequencies agree well with the ones observed in other experiments in para-H2 matrix (in the case of CO), and in Ar matrix, as well as in the gas phase (for HĊ=O (Tam & Fajardo 2001; Milligan & Jacox 1969; Sappey & Crosley 1990). This observation aligns well with the findings of Chen & Zhu (2003), who investigated the potential dissociation pathways of GO under different wavelengths. During UV irradiation within the range of 320–370 nm, they observed two distinct dissociation routes of GO: one resulting in the formation of HĊ=O + HĊ=O and the other leading to the dissociation products of HĊ=O + CO + H.
As the quantity of GO decreased under 365 nm UV irradiation, it can raise the question of whether using a higher wavelength of UV irradiation would be more suitable for generating Cl atoms. While Cl2 can only be photolyzed below 400 nm, GO undergoes decomposition even at higher wavelengths, up to 420 nm (Chen & Zhu 2003). Therefore, the change in the applied photolysis wavelength cannot help in this case. Although some GO decomposed upon 365 nm UV irradiation, the remaining amount was still sufficient to observe the GO + H reactions in the subsequent steps of H-atom formation and the dark period.
Upon analyzing the experimental results of the GO + H reaction, we identified both H-atom-abstraction and H-atom-addition reaction products. Among the reaction products of H-atom-abstraction from GO, we observed the formation of the R1 radical. We assign the bands with a correlation in their intensity change at 2082.0, 1451.3, and 1317.5 cm−1 to this species (see Fig. 4, and also Fig. S1 in the supporting information), as is supported by the close agreement between computed and observed vibrational frequencies and intensities (see Table 2). There are two other bands present in the close vicinity of the 2082.0 cm−1 band: one at 2078.6 cm−1 and another at 2084.0 cm−1, which might alternatively be assigned to R1. However, at the onset of 2217 nm NIR irradiation, when H atom formation began, their intensity increased slower than that of the 2082.0 cm−1 band. Later in the NIR irradiation, their intensity increased more rapidly. This indicates that the bands at 2078.6 cm−1 and 2084.0 cm−1 likely belong to complexes, such as HCl complexes of R1. Neither of these bands was present in the blank experiment, as is shown in Fig. S2 in the supporting information.
Changes in the quantity of the R1 radical during 2217 nm NIR irradiation and in the dark period are depicted in Fig. 5, in which the temporal evolution of the mixing ratios of the identified species is illustrated. These ratios were computed using the theoretical IR intensities by the method outlined by Tsuge & Lee (2020); our implementation is fully described elsewhere (Góbi et al. 2024). The formation of the R1 radical during 2217 nm NIR irradiation coincided with a decrease in the quantity of GO. During the dark period, the intensity of the R1 band decreased from 0.89 ppm to 0.67 ppm. This can be explained by either the abstraction of a second H atom from R1, as is discussed below, or by the recombination of R1 with an H atom. Together, these two reactions, which consume R1, occur faster than the H-atom-abstraction reaction from GO, forming R1.
In addition to the H-atom-abstraction reaction product, among the two possible H-atom-addition reactions, the addition of H to the O atom was observed in the experiments. This reaction resulted in the formation of syn,trans- and anti,trans-R2 radicals, identified by bands appearing at 1073.7 and 1230.5 cm−1, respectively. Similar to R1, these radicals were identified by comparing theoretically computed and experimentally observed vibrational frequencies, listed in Tables 3–4. These radicals did not form in the matrix upon 365 nm UV irradiation, but their quantity began to increase during 2217 nm NIR irradiation, coinciding with the decrease in the quantity of GO. Overnight, while the matrix was kept in the dark, the intensity of the bands corresponding to syn,trans-R2 decreased from 1.03 ppm to 0.92 ppm, while those of anti,trans-R2 increased from 0.26 ppm to 0.31 ppm. This can likely be explained by the fact that during the dark period and the 2217 nm irradiation event the rates of H-atom-abstraction reactions and recombination reactions of H atoms with R2 vary, and these relative rates differ among the different conformers.
In principle, a subsequent H-atom-addition reaction of either the syn,trans-R2 or the anti,trans-R1 radical would result in the production of GA. Although these reactions, being radical-radical recombination processes, are barrierless, the formation of GA was not observed in our experiments. This might be explained by competitive and faster H-atom-abstraction reactions of the R2 radicals.
The formation of the trans-R3, radical, which potentially represents the smallest barrier (see Table 1 and Fig. 3), was not detected during the experiments. It might be explained by a high reaction rate of the recombination to form GO + H2 or to form H2C=O + CO + H2.
The appearance of the R1 radical upon the H-atom reactions raises an interesting question. As was suggested by the previously described computations, in a barrierless reaction an H atom can recombine with the O atom that has a radical center in R1, resulting in the formation of HK. This molecule serves as a possible link between GO and GA, being an isomer of GO and a dehydrogenated form of GA. The theoretical computations predicted that the most intense band of the gauche-HK would be at 2146.1 cm−1 (see Table S67 in the supporting information). This is close to a band appearing during the NIR irradiation and in the dark at 2123.3 cm−1, but it is also close to the emerging intense bands of CO at 2143.2 cm−1, which might hide a weaker band. Unfortunately, due to the very low computed IR intensity of other vibrational modes of this molecule, these cannot be identified under our experimental conditions. Given these considerations, the formation of HK in our experiments is uncertain.
An H atom can also react with R1 in a barrierless H-atom-abstraction reaction, resulting in the formation of O=C=C=O, which is a metastable molecule and dissociates immediately into two CO molecules. Consistent with this, as was previously mentioned, the band of CO is discernible in the IR spectrum at 2143.2 cm−1. The CO quantity increased during NIR irradiation and throughout the dark period, reaching all in all 18.14 ppm (see Fig. 5).
Besides CO, the amount of HĊ=O also increased during the NIR irradiation to 6.63 ppm and slightly decreased in the dark to 6.19 ppm. Furthermore, the formation of H2C=O was observed through the appearance of its IR bands at 1498.1 and 1751.7 cm−1 during NIR irradiation, which closely match gas-phase values (Shimanouchi 1972). The quantity of H2C=O continued to rise in the dark to a final value of 3.37 ppm. These observations can be explained by consecutive H-atom addition to CO, which was observed in previous experiments (Hiraoka et al. 2002; Watanabe & Kouchi 2002).
To study the photophysical properties of the radicals (as it was mentioned in Sec. 2), a secondary UV irradiation was applied after the complete H-atom reaction cycle; that is, after the dark period, as is described in Sec. 2. The identified radicals, R1, syn,trans-R2, and anti,trans-R2, exhibited similar behavior under UV irradiation (at 300, 270, 240, and 216 nm wavelength), with their quantities continuously decreasing (see Fig. S3 in the supporting information). There were three instances in which the quantity of the radicals did not change (in the case of R1 at 240 and 216 nm irradiation, and in the case of syn,trans-R2 at 216 nm irradiation); however, this was due to the absence of any of the radicals left in the matrix at those specific times during the consecutive UV irradiation experiments. Regarding the decomposition products of CO, HĊ=O, and H2C=O, their quantities consistently increased during UV irradiation, except HĊ=O whose quantity decreased upon 240 and 216 nm irradiation (refer to Fig. S3 in the supporting information).
In conclusion, both H-atom-addition and H-atom-abstraction reactions were observed during the GO + H reaction, as is evidenced by the identification of R1, syn,trans-R2, and anti,trans-R2 radicals. A second H-atom-abstraction reaction of R1 results in the cleavage of the C−C bond, leading to the formation of two CO molecules. The formation of HK by subsequent H-atom-addition and H-atom-abstraction reactions cannot be definitively confirmed. Notably, each of the R1, syn,trans-R2, and anti,trans-R2 radicals is photolabile, decomposing upon UV irradiation.
 |
Fig. 1 Relative energies of the conformers of GO, GA, and EG, computed at the B3LYP/cc-pVTZ level of theory. The energies include zero-point vibrational energy (ZPVE) corrections. As the energy of the enol forms of GO and GA are much higher than that of the keto forms, these are not shown in the figure. |
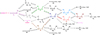 |
Fig. 2 Possible reactions of GO, GA, and EG with H atoms. The straight lines represent H-atom-addition reactions, while the dotted lines represent H-atom-abstraction reactions. The reactions denoted by colored lines have been observed experimentally. The figure only displays the species formed through one or two H-atom reactions of GO, GA, or EG to enhance clarity. Hydroxyketene, ketene, 1,2-ethenediol, vinyl alcohol, and acetaldehyde are denoted as HK, KE, ED, VA, and AA, respectively. CB1 and CB2 refer to carbenes. Additionally, R5, HK and KE are shown twice in the figure for better transparency. |
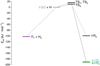 |
Fig. 3 Potential energy surfaces (PESs) for hydrogen atom reactions involving trans-GO, computed at the B3LYP/cc-pVTZ level of theory. R1: O=Ċ−CH=O, R2: HO−ĊH−CH=O R3: HC(O)−C(O)H2. The energies include ZPVE corrections. |
Relative energies of the H-atom reactions of trans-GO.
 |
Fig. 4 Overview spectra of the GO + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing trans-GO and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation, (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum, corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R1: O=Ċ−CH=O, R2: HO−ĊH−CH=O. |
Vibrational wave numbers and IR intensities of the O=Ċ−CH=O (R1) radical.
 |
Fig. 5 Temporal evolution of the mixing ratio of O=Ċ−CH=O (R1), syn,trans−HO−ĊH−CH=O (syn,trans-R2), anti,trans−HO−ĊH−CH=O (anti,trans-R2), CO, HĊ=O, and H2C=O the experiment of the GO + H reaction. The vertical lines refer to the end of the 2217 nm NIR irradiation, the horizontal lines refer to the quantity of the species at the end of the 2217 nm NIR irradiation. We note that some CO and HĊ=O were formed before the 2217 nm NIR irradiation, during the 365 nm photolysis. |
Vibrational wave numbers and IR intensities of syn,trans−HO−ĊH−CH (syn,trans-R2) radical.
Vibrational wave numbers and IR intensities of anti,trans−HO−ĊH−CH=O (anti,trans-R2) radical.
3.2 Glycolaldehyde (GA)
According to theory, four conformers of GA exist; namely syn,cis-, anti,cis-, gauche,trans-, and anti,trans-GA. Among these isomers, syn,cis-GA possesses the lowest energy, which can be explained by the intramolecular H-bond that stabilizes the molecular structure. The relative energies of these conformers are depicted in Fig. 1. This energy order is consistent with former quantum chemical computations (Chin et al. 2014). Similar to GO, GA has also a keto-enol tautomer, 1,2-ethenediol (ED, OH−CH=CH−OH), which exhibits five different conformers at the B3LYP/cc-pVTZ level of theory: anti,syn,cis-, anti,anti,cis-, syn,syn,trans-, anti,syn,trans-, and anti,anti,trans-ED. Their energies are higher than that of the most stable keto form (syn,cis-GA) by 33.4, 48.9, 48.5, 50.5, and 51.4 kJ mol−1, respectively.
In our experiments, after the deposition process, only syn,cis-GA was identified in the para-H2 matrix. The IR bands associated with syn,cis-GA are present in the IR spectrum at 3538.8, 1749.7, 1364.0, 1270.0, 1111.3, 859.6, and 750.9 cm−1. This observation aligns well with computations and is corroborated by earlier experiments conducted in N2, Ar, Ne, and para-H2 matrices (Chin et al. 2014; Duvernay et al. 2017). The recorded vibrational frequencies of GA and the previously documented values are detailed in Table S67 in the supporting information.
The GA + H reactions were examined theoretically and the results are briefly described here. The detailed computational results can be found in the supporting information (in Tables S68–S70) for the GA conformers that were not present in our experiments. The possible GA + H reactions are depicted in Fig. 2. Specifically, there are three potential sites for H-atom abstraction in the molecule: −CH, −CH2−, and −OH. The H-atom abstraction from −CH−, resulting in the formation of syn,cis−O=Ċ−CH2−OH (syn,cis-R4), exhibits a slight energy barrier of 0.5 kJ mol−1. The H-abstraction from the −CH2 group gives rise to the syn,cis−O=CH−ĊH−OH (syn,cis-R2) radical with an energy barrier of approximately 3.1 kJ mol−1. Lastly, the H-atom-abstraction reaction from the hydroxyl functional group to form cis−O=CH−CH2−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq13.png) (cis-R3) is predicted to have an energy barrier of 33.3 kJ mol−1. The computations performed by Álvarez-Barcia et al. (2018) produced results closely aligned with ours, with one notable exception: the barrier for the formation of the R2 radical. In this particular instance, their findings surpassed those derived from the B3LYP/cc-pVTZ level of theory.
(cis-R3) is predicted to have an energy barrier of 33.3 kJ mol−1. The computations performed by Álvarez-Barcia et al. (2018) produced results closely aligned with ours, with one notable exception: the barrier for the formation of the R2 radical. In this particular instance, their findings surpassed those derived from the B3LYP/cc-pVTZ level of theory.
Regarding the H-addition reactions, there are three possibilities: H-addition on the formyl O atom, on the formyl C atom, or on the hydroxyl group of the molecule. The H-addition on the formyl O atom exhibits a barrier of 39.4, and 37.4 kJ mol−1, while the H-addition on the C atom of the formyl group has a barrier of 30.8 kJ mol−1. These reactions lead to the formation of syn,syn,cis−HO−ĊH−CH2−OH (syn,syn,cis-R5), anti,syn,cis−HO−ĊH−CH2−OH (anti,syn,cis-R5), and syn,cis−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq14.png) −CH2−CH2−OH (syn,cis-R6), respectively. Lastly, the H-atom-addition on the −OH group results in bond cleavage, forming the O=CH−ĊH2 radical (R7) and a H2O molecule, with a barrier of 74.0 kJ mol−1. The PES of all the mentioned reactions can be seen in Fig. 6, the energy barriers of the reactions are listed in Table 5.
−CH2−CH2−OH (syn,cis-R6), respectively. Lastly, the H-atom-addition on the −OH group results in bond cleavage, forming the O=CH−ĊH2 radical (R7) and a H2O molecule, with a barrier of 74.0 kJ mol−1. The PES of all the mentioned reactions can be seen in Fig. 6, the energy barriers of the reactions are listed in Table 5.
Additionally, it is worth noting that GO and hydroxyketene (HK) can be formed through the abstraction of two H atoms, while EG can be formed via the addition of two H atoms of GA. In all of these cases, the second step of the reaction is barrierless. Therefore, the R2 and R3 radicals hold significance, as they serve as precursors of GO, whereas the R5 and R6 radicals can lead to the formation of EG.
Now, we shift the discussion to the experimental results: following the deposition of GA in solid para-H2, H atoms were generated in the matrix using the method detailed in Sec. 2. We employed 365 nm LED irradiation to generate Cl atoms, as no notable photodecomposition of GA upon 365 nm UV irradiation was detected in the blank experiments conducted in the absence of Cl2 in the matrix (see Fig. S4 in the supporting information).
During the GA + H reaction, a decrease in the quantity of GA was accompanied by the emergence of new spectral bands, attributed to the H-atom-abstraction reactions of GA. The abstraction of H atoms from the −CH2 – group of GA resulted in the formation of the syn,cis-R2 radical, as indicated by bands at 1575.4, 1176.2, 991.5, and 802.6 cm−1. Conversely, abstraction from the carbonyl C atom led to the generation of the syn,cis-R4 radical, evidenced by bands at 1867.8, 1046.9, 894.2, and 678.0 cm−1 in the spectrum (see Fig. 7, and also Fig. S5 in the supporting information). The identification of radicals syn,cis-R2 and syn,cis-R4 is based on computations. The positions of the aforementioned vibrational bands in the spectrum were in good agreement with the calculated vibrational frequencies, as is detailed in Tables 6 and 7. Besides the computations, the assignment of the syn,cis-R2 and syn,cis-R4 radicals is based on the fact that their quantity increased during the 365 nm UV and the 2217 nm NIR irradiation, as well as overnight in the dark, to reach a quantity of 3.43 ppm and 0.97 ppm, respectively (refer to Fig. 8). The identifications are further strengthened that among the possible GA + H reactions the formation of syn,cis-R2 and syn,cis-R4 have the smallest barriers (see Fig. 6).
In a second barrierless H-atom-abstraction reaction via the syn,cis-R2 or syn,cis-R4 radical, HK might be generated from GA. In our experiments, we observed the emergence of a band at 2123.9 cm−1 with very low intensity, which might be assigned to the HK molecule. Alternatively, as is discussed in Sect. 3.1, the intense band of CO 2143.2 cm−1 can hide the most intense band of HK. Since no other bands of HK could be identified, similar to the GO experiments, the formation of HK in the GA + H reaction cannot be confirmed or excluded.
With less confidence, an H-atom-addition reaction product of GA, R6, might also have been identified. A band that emerged during the 2217 nm radiation at 1088.7 cm−1 corresponds reasonably well with an intense band of R6 conformers (see Table 8). Unfortunately, we cannot assign further bands, as the bands computed with higher intensity either overlap with other species or are outside of our spectral region. Based on the computed IR spectra, either syn,cis-R6 or anti,cis-R6 could be assigned to the band at 1088.7 cm−1. Considering that the direct product of the syn,cis-GA + H reaction is syn,cis-R6, it is reasonable to infer that the bands correspond to the syn,cis-R6 conformer, especially given its low energy among the four possible conformers.
During the experiments, we observed the formation of other smaller molecular species; namely, CO, HĊ=O formaldehyde (H2C=O), and hydroxy methyl radical (H2Ċ−OH). CO can be identified at 2143.2 cm−1, HĊ=O at 1865.0 cm−1, H2C=O at 1498.1 cm−1, and H2Ċ−OH at 1179.4 cm−1. These assignments are consistent with previous experiments (Tam & Fajardo 2001; Milligan & Jacox 1969; Sappey & Crosley 1990; Shimanouchi 1972; Jacox 1981; Jacox & Milligan 1973), referring to Tables S65–S66 and S71–S72 in the supporting information. In detail, all these species are formed already under the 365 nm UV LED irradiation. During 2217 nm NIR irradiation, the bands of CO, H2Ċ−OH, and H2C=O showed increased intensity (with a final value of 2.72 ppm, 2.66 ppm, and 1.93 ppm, respectively), while the intensity of the band of HĊ=O decreased (to 1.46 ppm). Throughout the dark period, only the quantity of CO and H2C=O increased (to 3.40 ppm, and 4.85 ppm, respectively), while the quantities of the other two molecules decreased (in the case of HĊ=O to 1.20 ppm, in the case of H2Ċ−OH to 2.71 ppm). Since none of the mentioned four molecules formed in detectable amounts in the Cl2-free blank experiment under 365 nm irradiation (see Fig. S4 in the supporting information), it is reasonable to assume that their formation was facilitated by Cl atoms generated during the 365 nm UV irradiation (and consumed upon exposure to 2217 nm radiation, as is discussed in Sect. 2).
The Cl-atom-initiated gas-phase decomposition of GA was examined by Niki et al. (1987). Similarly, OH radical can also initiate the decomposition of GA (Niki et al. 1987; Butkovskaya et al. 2006; Bacher et al. 2001; Magneron et al. 2005). According to Niki et al. (1987) Cl atoms can abstract an H atom from GA, resulting in the formation of R4 radical. This reaction can explain the fact that R4 was formed not just during the 2217 nm NIR irradiation, but also during the 365 nm UV irradiation.
Another process, a second H-atom reaction, can also contribute to the formation of CO and H2C=O. According to the theoretical computations, R4 radical can take place in a second, barrierless H-atom-abstraction reaction which leads to the formation of O=Ċ−CH2−O. This radical is not stable, it decomposes to CO and H2C=O. This process can account for the observed increase in the quantity of CO and H2C=O not only during 365 nm and 2217 nm NIR irradiations but also during the dark period. Another potential pathway for CO and H2C=O formation is through the HĊ=O + HĊ=O reaction, which results in the production of these two molecules. This process can explain the decrease in HĊ=O quantity observed during 2217 nm NIR irradiation, as it facilitates the diffusion of HĊ=O species.
After investigating the H-atom reactions of GA, secondary UV photolysis was applied, as is detailed in Sect. 2. Under 300, 270, 240, and 216 nm irradiation, the quantities of syn,cis-R2 and syn,cis-R4 radicals generally decreased, with two exceptions: the quantity of R4 slightly increased (possibly due to photoinduced tautomerization of R2) under 300 nm irradiation, and its quantity remained unchanged during 270 nm irradiation (see Fig. S6 in the supporting information). The decrease in the intensity of the bands of the radicals indicates their instability under UV irradiation. This aligns with the overall increase in the quantities of CO, HĊ=O, H2Ċ−OH and H2C=O during the secondary UV irradiation. Specifically, the bands of CO increased at every used wavelength. The quantity of HĊ=O increased during 300, 270, and 240 nm irradiation but decreased upon 216 nm irradiation. The intensity of the band of H2Ċ−OH remained unchanged during 300 nm irradiation, decreased during 270 nm irradiation, and increased during 240 and 216 nm irradiation. Finally, the quantity of H2C=O decreased during 300 nm irradiation and increased during 270, 240, and 216 nm irradiations.
In conclusion, the H-abstraction reaction of GA was observed through the formation of the syn,cis-R2 and syn,cis-R4 radicals. With a degree of uncertainty, syn,cis-R6 and HK were also tentatively identified.
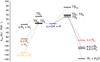 |
Fig. 6 Potential energy surfaces (PESs) for hydrogen atom reactions involving syn,cis-GA, computed at the B3LYP/cc-pVTZ level of theory. R2: O=CH−ĊH−OH, R3: to O=CH−CH2−O⋅, R4: O=Ċ−CH2−OH, R5: HO−ĊH−CH2−OH, R6: |
![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq15.png)
Relative energies of the H-atom reactions of syn,cis-GA.
 |
Fig. 7 Overview spectra of the GA + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing syn,cis-GA and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation, (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R2: O=CH−ĊH−OH, R4: O=Ċ−CH2−OH. |
Vibrational wave numbers and IR intensities of syn,cis−O=CH−ĊH−OH (syn,cis-R2) radical.
Vibrational wave numbers and IR intensities of syn,cis-O=Ċ−CH2−OH (syn,cis-R4) radical.
 |
Fig. 8 Temporal evolution of the mixing ratio of syn,cis−HO−ĊH−CH=O (syn,cis-R2), syn,cis−O=Ċ−CH2−OH (syn,cis-R4), |
![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq21.png)
Vibrational wave numbers and IR intensities of syn,cis−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq22.png) −CH2−CH2−OH (syn,cis-R6) radical.
−CH2−CH2−OH (syn,cis-R6) radical.
3.3 Ethylene glycol (EG)
In line with former studies (Christen et al. 2001; Guo et al. 2018), six conformers of EG could be identified by computations. The minimum energy structure is the anti,syn,cis-EG conformer, as this structure offers the potential for intramolecular H-bond formation. Anti,syn,cis-EG, as well as the structure and the relative energies of the other five identified conformers; namely, syn,gauche,cis-, anti,gauche,cis-, gauche,gauche,trans-, anti,gauche,trans-, and anti,anti,trans-EG are shown in Fig. 1. In the deposited para-H2 matrix, only one conformer of EG was identified, specifically the anti,syn,cis-EG. Its key bands are observed in the IR spectrum at 3624.1, 2968.7, 2948.8, 2934.7, 2929.5, 2887.1, 1470.1, 1462.8, 1385.8, 1348.2, 1267.0, 1242.4, 1161.3, 1101.1, 1070.7, 1043.0, 880.2, and 867.6 cm−1. This identification aligns not only with theory but also with previous experiments conducted in Ar and Xe matrices (Frei et al. 1977). The observed bands of the sample molecule, along with previously documented vibrational frequencies in noble gas matrices, are presented in Table S74 of the supporting information.
The EG + H reactions were computationally investigated, similar to the approaches taken for GO and GA. The predicted reaction pathways were explored for all conformers of EG, and here we present data specifically for the conformer observed in our experiments. Additional computed data for the other isomers is available in the supporting information in Tables S75–S79. Figure 2 illustrates the possible EG + H reactions. The H-atom-abstraction channel involves the −CH2− and −OH reactive sites of EG, resulting in the formation of anti,syn,cis−HO−ĊH−CH2−OH (anti,syn,cis-R5) and anti,syn,cis−![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq25.png) −CH2−CH2−OH (anti,syn,cis-R6) radicals, with theoretical barriers of 4.2 kJ mol−1 and 22.5 kJ mol−1, respectively. Due to the saturation of C atoms in EG, the only viable H-atom-addition reaction is the addition to the hydroxyl group. This reaction involves an energy barrier of 80.0 kJ mol−1 and leads to the breaking of the C−O bond, resulting in the formation of ĊH2−CH2−OH (R8) radical and H2O. The PESs of the plausible H-atom-abstraction and H-atom-addition reactions of EG are depicted in Fig. 9, and the energy barriers of the reactions are listed in Table 9. The computational results of Álvarez-Barcia et al. (2018) correlate well with ours in the case of the R6 radical, but we obtained a considerably lower energy barrier for the formation of the R5 radical.
−CH2−CH2−OH (anti,syn,cis-R6) radicals, with theoretical barriers of 4.2 kJ mol−1 and 22.5 kJ mol−1, respectively. Due to the saturation of C atoms in EG, the only viable H-atom-addition reaction is the addition to the hydroxyl group. This reaction involves an energy barrier of 80.0 kJ mol−1 and leads to the breaking of the C−O bond, resulting in the formation of ĊH2−CH2−OH (R8) radical and H2O. The PESs of the plausible H-atom-abstraction and H-atom-addition reactions of EG are depicted in Fig. 9, and the energy barriers of the reactions are listed in Table 9. The computational results of Álvarez-Barcia et al. (2018) correlate well with ours in the case of the R6 radical, but we obtained a considerably lower energy barrier for the formation of the R5 radical.
There are two important radicals among the possible products of the EG + H reaction; namely, R5 and R6. They can be the connection link between EG and GA, as GA can be formed from R5 or from R6 in a barrierless reaction, or the 1,2-ethenediol (ED, OH−CH=CH−OH) can be formed from R5, also in a barrierless reaction (see these reactions in Fig. 2).
As in the cases of GO and GA, H atoms were generated in the para-H2 matrix after deposition, and the EG + H reactions were investigated. Upon H-atom generation, two bands appeared in the spectrum at wave numbers 1144.8 and 1219.9 cm−1 (see Fig. 10, and also Fig. S7 in the supporting information). These bands were not present after the 365 nm UV irradiation; they started to appear during the 2217 nm NIR irradiation. The assignment of the 1144.8 cm−1 band is not clear; according to theoretical computations, this band may arise from the presence of either anti,syn,cis-R5 or anti,anti,cis-R5 radicals. Notably, anti,syn,cis-R5 and anti,anti,cis-R5 only differ in the conformation of the hydroxyl group attached to the radical C atom. Since the anti,syn,cis conformer can be formed directly from the anti,syn,cis-EG molecule, we might tentatively assign this band to this species. Unfortunately, no other bands can be assigned due to the overlap with the bands of EG.
Based on computed IR spectra, the band at 1219.9 cm−1 can be most likely assigned to anti,anti,cis-R5. The formation of this radical requires a conformational change around the −OH-groups, which could occur due to the dissipation of the energy produced by the EG + H reaction. Accepting this assignment, this conformer could also be attributed to the 1144.8 cm−1 band, or at least it may contribute to it.
The intensity of the band at 1219.9 cm−1 increased not only during NIR irradiation but also during the dark period, reinforcing that it can be assigned to products of an H-atom reaction (see Fig. 11). In contrast, the intensity of the band at 1144.8 cm−1 fluctuated overnight. This could be explained either by a competing recombination reaction with H atoms or by the possible spectral overlap of this band. All in all, to the end of the dark period the quantity of anti,syn,cis-R5 radical reached 0.68 ppm, and the quantity of anti,anti,cis-R5 radical reached 5.77 ppm.
Although anti,syn,cis-R5 and anti,anti,cis-R5 could be identified only based on the good correlation between one and two computed and experimental frequencies (Tables 10 and 11), as was already discussed, the formation of R5 has the smallest barrier, 4.2 kJ mol−1, among the possible EG + H reactions, supporting the identification of these radicals.
During H-atom generation, not only did the bands of R5 conformers emerge in the spectrum, but the presence of two additional molecules–formaldehyde (H2C=O) and hydroxy methyl radical (H2Ċ−OH)-was also identified ((Shimanouchi 1972; Jacox 1981; Jacox & Milligan 1973), see Tables S72–S73 in the supporting information). None of the mentioned radicals, H2C=O, or H2Ċ−OH were present in the blank experiments (referring to Fig. S8 in the supporting information). However, the quantities of H2C=O and H2Ċ−OH increased significantly during the 365 nm irradiation. As is detailed in Sect. 2, H atoms were generated in a two-step process, where Cl atoms were formed initially through the 365 nm UV photodecomposition of Cl2. This suggests that the formation of the two molecules is not linked to the presence of H atoms but rather to the presence of Cl atoms.
During secondary UV irradiation, which aimed to support the assignment of the bands (see Sect. 2), the intensity of the bands assigned to the radicals remained unchanged upon 300 nm irradiation but decreased during 270, 240, and 216 nm irradiation (refer to Fig. S9 in the supporting information). This indicates their instability under UV irradiation. At this experimental stage, the quantity of H2C=O increased at every applied wavelength, while the intensity of the H2Ċ−OH band exhibited variation: it increased upon 300 nm irradiation, decreased during 270 nm irradiation, remained unchanged at 240 nm, and increased again upon 216 nm irradiation. Two other small molecules – namely, CO and HĊ=O (with bands at positions 2143.4 and 1864.7 cm−1, respectively) – appeared in the spectrum during secondary UV irradiation.
In conclusion, EG undergoes H-atom-abstraction reactions with H atoms. However, due to the spectral congestion resulting from the rich spectrum of EG and the potential presence of multiple conformers with similar IR spectra among the possible products, drawing conclusive results from the experiments is challenging compared to those with GO and GA. Among the reaction products, both anti,syn,cis-R5 and anti,anti,cis-R5 could be tentatively assigned. However, both decompose when irradiated with UV light of 270 nm or shorter wavelengths.
 |
Fig. 9 Potential energy surfaces (PESs) for hydrogen atom reactions involving anti,syn,cis-EG, computed at the B3LYP/cc-pVTZ level of theory. R5: HO−ĊH−CH2−OH, R6: |
![Mathematical equation: $\[\dot{\mathrm{O}}\]$](/articles/aa/full_html/2024/09/aa50649-24/aa50649-24-eq26.png)
Relative energies of the H-atom reactions of anti,syn,cis-EG.
 |
Fig. 10 Overview spectra of the EG + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing anti,syn,cis-EG and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation. (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R5 refers to HO−ĊH−CH2−OH. |
 |
Fig. 11 Temporal evolution of the mixing ratio of HO−ĊH−CH2−OH (R5), H2C=O, and H2Ċ−OH and H2Ċ−OH in the experiment of EG + H reaction. The vertical lines refer to the end of the 2217 nm NIR irradiation, the horizontal lines refer to the quantity of the species at the end of the 2217 nm NIR irradiation. |
Vibrational wave numbers and IR intensities of anti,syn,cis−HO−ĊH−CH2−OH (anti,syn,cis-R5) radical.
Vibrational wave numbers and IR intensities of anti,anti,cis−HO−ĊH−CH2−OH (anti,anti,cis-R5) radical.
4 Conclusions
Glyoxal (GO), glycolaldehyde (GA), and ethylene glycol (EG) could potentially act as interstellar precursors for larger sugar molecules if activated by either UV or cosmic radiation or by reactions involving higher-energy radicals. To investigate this hypothesis, we explored the H-atom reactions of these species in a 3.1 K solid para-H2 matrix, simulating apolar ice environments.
While the solid para-H2 matrix environment employed in our study differs from typical interstellar ice environments in terms of polarity and temperature (maintained at <4 K), if a reaction occurs under our investigated conditions, it is likely to remain open under astrophysical conditions with temperatures exceeding 10 K. While there may be variations in reaction rates, including those of H-atom-addition and H-atom-abstraction processes, at slightly higher temperatures, our method is valuable for evaluating the feasibility of reactions at low temperatures. It allows for assessing tunneling or barrierless processes and offers direct comparisons with quantum chemical computations for isolated molecules. With sharp, well-resolved vibrational bands ensuring reliable assignments and the straightforward deduction of reaction mechanisms, our method boasts high sensitivity facilitated by a large mixing ratio of H atoms, quantum diffusion within the para-H2 matrix, and a high signal-to-noise ratio. Thus, it allows for studying slow processes on a laboratory timescale.
In a former study with GO ice, the reaction of H atoms with GO led to its decomposition (Leroux et al. 2020). In our present study, we observed this channel, resulting in the formation of CO via the O=Ċ−CH=O (R1) radical in a two-step H-atom-abstraction process. Additionally, compared to the former GO ice study, our current research allowed for the identification of minor products; namely, syn,trans−HO−ĊH−CH=O (syn,trans-R2) and anti,trans−HO−ĊH−CH=O (anti,trans-R2), generated by H-atom-addition processes. In the H-atom abstraction 9.74 ppm GO participated, while the total amount of H-atom-addition product was 1.23 ppm at the end of the dark period of the experiment. Although not identified unambiguously, O=C=CH−OH (HK) might also form through H-atom-assisted tautomerization. We did not observe the formation of GA from GO in H-atom reactions, possibly due to more rapid H-atom-abstraction reactions. The observed species are listed in Table 12.
In contrast to experiments conducted with GA ice (Leroux et al. 2021), H-atom reactions did not lead to the formation of EG, the product resulting from the addition of two H atoms to GA. In our present study, we primarily observed H-atom-abstraction products, specifically syn,cis−O=CH−ĊH−OH(syn,cis-R2) and syn,cis−O=Ċ−CH2−OH (syn,cis-R4) radicals (see Table 12), in a total quantity of 4.40 ppm. Additionally, we tentatively identified HK, which is a product of two H-atom abstractions from GA. This discrepancy in results may be attributed to differences between apolar and polar ice environments, variations in temperature, or possibly a higher flux of H atoms in the former experiment. In our current experiment, only a single H-atom-addition product, syn,cis-R6, was tentatively identified (referring to Table 12); at the end of the H-atom reaction it was present in 3.52 ppm.
Regarding EG, the present study revealed that H-atom-abstraction reactions are feasible at low temperatures. Anti,syn,cis−HO−ĊH−CH2−OH (anti,syn,cis-R5) and anti,anti,cis−HO−ĊH−CH2−OH (anti,anti,cis-R5) were tentatively assigned (see Table 12); their total amount was 6.45 ppm in the matrix at the end of the H-atom reaction.
Species observed from H-atom reactions during each experiment.
5 Astrophysical implications
The present study demonstrates that H-atom-abstraction and H-atom-addition reactions at low temperatures can lead to the formation of radicals from GO, GA, and EG in apolar ices. The reactions observed in our study suggest that similar processes could occur in cold prestellar cores, where H atoms are present but UV radiation is limited. These radicals can play a key role in interstellar sugar formation; for example, they can recombine in a barrierless process, resulting in larger sugar molecules. The identification of specific radicals and reaction products under these conditions provides valuable data for understanding chemical pathways in interstellar ice environments. Additionally, we observed H-atom-assisted reactions that lead to the destruction of molecules, indicating that the synthesis and the destruction of sugars are competitive processes. Furthermore, our findings contribute to understanding the complexity of chemical processes in astrophysical environments and highlight the importance of laboratory studies, including para-H2 matrix isolation studies, for exploring diverse reaction pathways.
Data availability
The data underlying this article are available in the article and in its online supplementary material (https://zenodo.org/records/12210022).
Acknowledgements
The authors would like to thank the Hungarian Academy of Sciences for funding this research through the Lendület program. The Hungarian Scientific Research Fund (OTKA K143196) and the NKFIH excellence grant TKP2021-NKTA-64 are also acknowledged. B.K. and A.S. were supported by the “ÚNKP-23-3 New National Excellence Program” of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund. B.K. was also supported by the “DKOP-23 Doctoral Excellence Program” of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund. S.G. was supported by the “Bolyai Ösztöndíj” of the Hungarian Academy of Sciences and by the “ÚNKP-23-5 New National Excellence Program” of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund.
References
- Ahmad, A., Shivani, Misra, A., & Tandon, P. 2020, Res. Astron. Astrophys., 20, 014 [CrossRef] [Google Scholar]
- Álvarez-Barcia, S., Russ, P., Kästner, J., & Lamberts, T. 2018, MNRAS, 479, 2007 [Google Scholar]
- Aviles-Moreno, J.-R., Demaison, J., & Huet, T. R. 2006, J. Am. Chem. Soc., 128, 10467 [CrossRef] [Google Scholar]
- Bacher, C., Tyndall, G. S., & Orlando, J. J. 2001, J. Atmos. Chem., 39, 171 [NASA ADS] [CrossRef] [Google Scholar]
- Bazsó, G., Csonka, I. P., Góbi, S., & Tarczay, G. 2021, Rev. Sci. Instrum., 92, 124104 [CrossRef] [Google Scholar]
- Beltrán, M. T., Codella, C., Viti, S., Neri, R., & Cesaroni, R. 2009, ApJ, 690, L93 [Google Scholar]
- Bennett, C. J., & Kaiser, R. I. 2007, ApJ, 661, 899 [Google Scholar]
- Biver, N., Bockelée-Morvan, D., Moreno, R., et al. 2015, Sci. Adv., 1, e1500863 [Google Scholar]
- Bock, C. W., Panchenko, Y. N., & Krasnoshchiokov, S. V. 1988, Chem. Phys., 125, 63 [CrossRef] [Google Scholar]
- Butkovskaya, N. I., Pouvesle, N., Kukui, A., & Le Bras, G. 2006, J. Phys. Chem. A, 110, 13492 [NASA ADS] [CrossRef] [Google Scholar]
- Butlerow, A. 1861, Justus Liebigs Ann. Chem., 120, 295 [CrossRef] [Google Scholar]
- Butscher, T., Duvernay, F., Theule, P., et al. 2015, MNRAS, 453, 1587 [NASA ADS] [CrossRef] [Google Scholar]
- Butscher, T., Duvernay, F., Rimola, A., Segado-Centellas, M., & Chiavassa, T. 2017, Phys. Chem. Chem. Phys., 19, 2857 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, Y., & Zhu, L. 2003, J. Phys. Chem. A, 107, 4643 [NASA ADS] [CrossRef] [Google Scholar]
- Chin, W., Chevalier, M., Thon, R., et al. 2014, J. Chem. Phys., 140, 224319 [NASA ADS] [CrossRef] [Google Scholar]
- Christen, D., Coudert, L. H., Larsson, J. A., & Cremer, D. 2001, J. Mol. Spectrosc., 205, 185 [NASA ADS] [CrossRef] [Google Scholar]
- Chuang, K.-J., Fedoseev, G., Ioppolo, S., Van Dishoeck, E. F., & Linnartz, H. 2016, MNRAS, 455, 1702 [NASA ADS] [CrossRef] [Google Scholar]
- Coutens, A., Persson, M. V., Jørgensen, J. K., Wampfler, S. F., & Lykke, J. M. 2015, A&A, 576, A5 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Coutens, A., Viti, S., Rawlings, J., et al. 2018, MNRAS, 475, 2016 [NASA ADS] [CrossRef] [Google Scholar]
- Crovisier, J., Bockelée-Morvan, D., Biver, N., et al. 2004, A&A, 418, L35 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- De Simone, M., Codella, C., Testi, L., et al. 2017, A&A, 599, 121 [Google Scholar]
- Duvernay, F., Butscher, T., Chiavassa, T., & Coussan, S. 2017, Chem. Phys., 496, 9 [CrossRef] [Google Scholar]
- Eckhardt, A. K., Bergantini, A., Singh, S. K., Schreiner, P. R., & Kaiser, R. I. 2019, Angew. Chem., Int. Ed., 58, 5663 [CrossRef] [Google Scholar]
- Engdahl, A., & Nelander, B. 1988, Chem. Phys. Lett., 148, 264 [CrossRef] [Google Scholar]
- Fedoseev, G., Cuppen, H. M., Ioppolo, S., Lamberts, T., & Linnartz, H. 2015, MNRAS, 448, 1288 [NASA ADS] [CrossRef] [Google Scholar]
- Fedoseev, G., Chuang, K.-J., Ioppolo, S., et al. 2017, ApJ, 842, 52 [NASA ADS] [CrossRef] [Google Scholar]
- Frei, H., Ha, T.-K., Meyer, R., & Gūnthard, H. H. 1977, Chem. Phys., 25, 271 [CrossRef] [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. 2009, Gaussian 09 Revision 71 A.2 (Wallingford, CT: Gaussian Inc.) [Google Scholar]
- Góbi, S., Csonka, I. P., Bazsó, G., & Tarczay, G. 2021, ACS Earth Space Chem., 5, 1180 [CrossRef] [Google Scholar]
- Góbi, S., Keresztes, B., Schneiker, A., Ragupathy, G., & Tarczay, G. 2024, J. Chem. Phys., 160, 024310 [CrossRef] [Google Scholar]
- Goesmann, F., Rosenbauer, H., Bredehöft, J. H., et al. 2015, Science, 349, aab0689 [Google Scholar]
- Guo, Y.-C., Cai, C., & Zhang, Y.-H. 2018, AIP Adv., 8, 055308 [NASA ADS] [CrossRef] [Google Scholar]
- Halfen, D. T., Apponi, A. J., Woolf, N., Polt, R., & Ziurys, L. M. 2006, ApJ, 639, 237 [CrossRef] [Google Scholar]
- Hiraoka, K., Sato, T., Sato, S., et al. 2002, ApJ, 577, 265 [Google Scholar]
- Hoffman, I. M., Goss, W. M., & Palmer, P. 2007, ApJ, 654, 971 [NASA ADS] [CrossRef] [Google Scholar]
- Hollis, J. M., Lovas, F. J., & Jewell, P. R. 2000, ApJ, 540, L107 [Google Scholar]
- Hollis, J. M., Lovas, F. J., Jewell, P. R., & Coudert, L. H. 2002, ApJ, 571, L59 [NASA ADS] [CrossRef] [Google Scholar]
- Houghton, S., & Whiteoak, J. 1995, MNRAS, 273, 1033 [NASA ADS] [CrossRef] [Google Scholar]
- Jacox, M. E. 1981, Chem. Phys., 59, 213 [CrossRef] [Google Scholar]
- Jacox, M. E., & Milligan, D. E. 1973, J. Mol. Spectrosc., 47, 148 [NASA ADS] [CrossRef] [Google Scholar]
- Jørgensen, J. K., Favre, C., Bisschop, S. E., et al. 2012, ApJ, 757, L4 [Google Scholar]
- Keresztes, B., Góbi, S., Csonka, I. P., et al. 2023, MNRAS, 521, 2649 [CrossRef] [Google Scholar]
- Kettwich, S. C., Raston, P. L., & Anderson, D. T. 2009, J. Phys. Chem. A, 113, 7621 [NASA ADS] [CrossRef] [Google Scholar]
- Khomenko, T. I., Sakharov, M. M., & Golovina, O. A. 1980, Russ. Chem. Rev., 49, 570 [NASA ADS] [CrossRef] [Google Scholar]
- Kleimeier, N. F., Eckhardt, A. K., & Kaiser, R. I. 2021, J. Am. Chem. Soc., 143, 14009 [CrossRef] [Google Scholar]
- Layssac, Y., Gutiérrez-Quintanilla, A., Chiavassa, T., & Duvernay, F. 2020, MNRAS, 496, 5292 [NASA ADS] [CrossRef] [Google Scholar]
- Lefloch, B., Ceccarelli, C., Codella, C., et al. 2017, MNRAS Lett., 469, L73 [NASA ADS] [CrossRef] [Google Scholar]
- Leroux, K., Guillemin, J.-C., & Krim, L. 2020, MNRAS, 491, 289 [NASA ADS] [CrossRef] [Google Scholar]
- Leroux, K., Guillemin, J.-C., & Krim, L. 2021, MNRAS, 507, 2632 [CrossRef] [Google Scholar]
- Li, Y., Si, D., Shang, W., et al. 2022, New J. Chem., 46, 6360 [CrossRef] [Google Scholar]
- Magneron, I., Mellouki, A., Le Bras, G., et al. 2005, J. Phys. Chem. A, 109, 4552 [NASA ADS] [CrossRef] [Google Scholar]
- Maity, S., Kaiser, R. I., & Jones, B. M. 2015, Phys. Chem. Chem. Phys., 17, 3081 [NASA ADS] [CrossRef] [Google Scholar]
- Mardyukov, A., Wende, R. C., & Schreiner, P. R. 2023, Chem. Commun., 59, 2596 [CrossRef] [Google Scholar]
- Matthews, D. A., Cheng, L., Harding, M. E., et al. 2020, J. Chem. Phys., 152, 214108 [NASA ADS] [CrossRef] [Google Scholar]
- Maury, A. J., Belloche, A., André, P., et al. 2014, A&A, 563, L2 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Melosso, M., Bizzocchi, L., Gazzeh, H., et al. 2022, Chem. Commun., 58, 2750 [NASA ADS] [CrossRef] [Google Scholar]
- Milligan, D. E., & Jacox, M. E. 1969, J. Chem. Phys., 51, 277 [NASA ADS] [CrossRef] [Google Scholar]
- Mizuno, T., & Weiss, A. H. 1974, in Advances in Carbohydrate Chemistry and Biochemistry, 29 (Elsevier), 173 [CrossRef] [Google Scholar]
- Niki, H., Maker, P. D., Savage, C. M., & Hurley, M. D. 1987, J. Phys. Chem., 91, 2174 [CrossRef] [Google Scholar]
- Orgel, L. E. 1998, TiBS, 23, 491 [Google Scholar]
- Paiva, M. A., Pilling, S., Mendoza, E., Galvão, B. R., & De Abreu, H. A. 2023, MNRAS, 519, 2518 [Google Scholar]
- Pantaleone, S., Enrique-Romero, J., Ceccarelli, C., et al. 2020, ApJ, 897, 56 [NASA ADS] [CrossRef] [Google Scholar]
- Raston, P. L., & Anderson, D. T. 2006, Phys. Chem. Chem. Phys., 8, 3124 [NASA ADS] [CrossRef] [Google Scholar]
- Raston, P. L., & Anderson, D. T. 2007, J. Chem. Phys., 126, 021106 [NASA ADS] [CrossRef] [Google Scholar]
- Raston, P. L., Kettwich, S. C., & Anderson, D. T. 2010, Low Temp. Phys., 36, 392 [NASA ADS] [CrossRef] [Google Scholar]
- Raston, P. L., Kettwich, S. C., & Anderson, D. T. 2015, J. Mol. Spectrosc., 310, 72 [NASA ADS] [CrossRef] [Google Scholar]
- Rivilla, V. M., Colzi, L., Jiménez-Serra, I., et al. 2022, ApJ, 929, L11 [NASA ADS] [CrossRef] [Google Scholar]
- Sappey, A. D., & Crosley, D. R. 1990, J. Chem. Phys., 93, 7601 [NASA ADS] [CrossRef] [Google Scholar]
- Schneiker, A., Góbi, S., Joshi, P. R., et al. 2021, J. Phys. Chem. Lett., 12, 6744 [CrossRef] [Google Scholar]
- Schneiker, A., Ragupathy, G., Bazsó, G., & Tarczay, G. 2022, J. Phys. Chem. A, 126, 2832 [NASA ADS] [CrossRef] [Google Scholar]
- Shimanouchi, T. 1972, National Standard Reference Data System, 39, Tables of molecular vibrational frequencies (Washington, DC: National Bureau of Standards) [Google Scholar]
- Silva, S. G., Vichietti, R. M., Haiduke, R. L., Machado, F. B., & Spada, R. F. 2020, MNRAS, 497, 4486 [NASA ADS] [CrossRef] [Google Scholar]
- Skouteris, D., Balucani, N., Ceccarelli, C., et al. 2018, ApJ, 854, 135 [NASA ADS] [CrossRef] [Google Scholar]
- Sorrell, W. H. 2001, ApJ, 555, L129 [NASA ADS] [CrossRef] [Google Scholar]
- Tam, S., & Fajardo, M. E. 2001, J. Low Temp. Phys., 122, 345 [CrossRef] [Google Scholar]
- Tsuge, M., & Lee, Y.-P. 2020, in Molecular and Laser Spectroscopy, Advances and Applications, 2, eds. V. Gupta, & Y. Ozaki (Amsterdam, Netherlands: Elsevier), 167 [Google Scholar]
- Watanabe, N., & Kouchi, A. 2002, ApJ, 571, L173 [Google Scholar]
- Woods, P. M., Kelly, G., Viti, S., et al. 2012, ApJ, 750, 19 [NASA ADS] [CrossRef] [Google Scholar]
- Woods, P. M., Slater, B., Raza, Z., et al. 2013, ApJ, 777, 90 [Google Scholar]
All Tables
Vibrational wave numbers and IR intensities of syn,trans−HO−ĊH−CH (syn,trans-R2) radical.
Vibrational wave numbers and IR intensities of anti,trans−HO−ĊH−CH=O (anti,trans-R2) radical.
Vibrational wave numbers and IR intensities of syn,cis−O=CH−ĊH−OH (syn,cis-R2) radical.
Vibrational wave numbers and IR intensities of syn,cis-O=Ċ−CH2−OH (syn,cis-R4) radical.
Vibrational wave numbers and IR intensities of syn,cis−−CH2−CH2−OH (syn,cis-R6) radical.
Vibrational wave numbers and IR intensities of anti,syn,cis−HO−ĊH−CH2−OH (anti,syn,cis-R5) radical.
Vibrational wave numbers and IR intensities of anti,anti,cis−HO−ĊH−CH2−OH (anti,anti,cis-R5) radical.
All Figures
|
Fig. 1 Relative energies of the conformers of GO, GA, and EG, computed at the B3LYP/cc-pVTZ level of theory. The energies include zero-point vibrational energy (ZPVE) corrections. As the energy of the enol forms of GO and GA are much higher than that of the keto forms, these are not shown in the figure. |
| In the text | |
|
Fig. 2 Possible reactions of GO, GA, and EG with H atoms. The straight lines represent H-atom-addition reactions, while the dotted lines represent H-atom-abstraction reactions. The reactions denoted by colored lines have been observed experimentally. The figure only displays the species formed through one or two H-atom reactions of GO, GA, or EG to enhance clarity. Hydroxyketene, ketene, 1,2-ethenediol, vinyl alcohol, and acetaldehyde are denoted as HK, KE, ED, VA, and AA, respectively. CB1 and CB2 refer to carbenes. Additionally, R5, HK and KE are shown twice in the figure for better transparency. |
| In the text | |
|
Fig. 3 Potential energy surfaces (PESs) for hydrogen atom reactions involving trans-GO, computed at the B3LYP/cc-pVTZ level of theory. R1: O=Ċ−CH=O, R2: HO−ĊH−CH=O R3: HC(O)−C(O)H2. The energies include ZPVE corrections. |
| In the text | |
|
Fig. 4 Overview spectra of the GO + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing trans-GO and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation, (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum, corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R1: O=Ċ−CH=O, R2: HO−ĊH−CH=O. |
| In the text | |
|
Fig. 5 Temporal evolution of the mixing ratio of O=Ċ−CH=O (R1), syn,trans−HO−ĊH−CH=O (syn,trans-R2), anti,trans−HO−ĊH−CH=O (anti,trans-R2), CO, HĊ=O, and H2C=O the experiment of the GO + H reaction. The vertical lines refer to the end of the 2217 nm NIR irradiation, the horizontal lines refer to the quantity of the species at the end of the 2217 nm NIR irradiation. We note that some CO and HĊ=O were formed before the 2217 nm NIR irradiation, during the 365 nm photolysis. |
| In the text | |
|
Fig. 6 Potential energy surfaces (PESs) for hydrogen atom reactions involving syn,cis-GA, computed at the B3LYP/cc-pVTZ level of theory. R2: O=CH−ĊH−OH, R3: to O=CH−CH2−O⋅, R4: O=Ċ−CH2−OH, R5: HO−ĊH−CH2−OH, R6: |
| In the text | |
|
Fig. 7 Overview spectra of the GA + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing syn,cis-GA and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation, (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R2: O=CH−ĊH−OH, R4: O=Ċ−CH2−OH. |
| In the text | |
|
Fig. 8 Temporal evolution of the mixing ratio of syn,cis−HO−ĊH−CH=O (syn,cis-R2), syn,cis−O=Ċ−CH2−OH (syn,cis-R4), |
| In the text | |
|
Fig. 9 Potential energy surfaces (PESs) for hydrogen atom reactions involving anti,syn,cis-EG, computed at the B3LYP/cc-pVTZ level of theory. R5: HO−ĊH−CH2−OH, R6: |
| In the text | |
|
Fig. 10 Overview spectra of the EG + H experiment at 3.1 K. (a) MIR spectrum recorded after depositing a para-H2 matrix containing anti,syn,cis-EG and Cl2 at 3.1 K. (b) Difference spectrum corresponding to the 365 nm irradiation. (c) Difference spectrum corresponding to the 2217 nm irradiation. (d) Difference spectrum corresponding to the dark process. (e) Difference spectrum corresponding to the second 365 nm irradiation and the second 2217 nm irradiation. Difference spectra were obtained by subtracting the MIR spectrum recorded before each experimental step. R5 refers to HO−ĊH−CH2−OH. |
| In the text | |
|
Fig. 11 Temporal evolution of the mixing ratio of HO−ĊH−CH2−OH (R5), H2C=O, and H2Ċ−OH and H2Ċ−OH in the experiment of EG + H reaction. The vertical lines refer to the end of the 2217 nm NIR irradiation, the horizontal lines refer to the quantity of the species at the end of the 2217 nm NIR irradiation. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.