| Issue |
A&A
Volume 699, July 2025
|
|
|---|---|---|
| Article Number | A235 | |
| Number of page(s) | 10 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202555360 | |
| Published online | 11 July 2025 | |
Interstellar chemistry of CN radicals on ices: The formation of CH3CN and CH3NC and potential connection to acetamide
1 Leiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University,
PO Box 9502,
2300 RA Leiden, The Netherlands
2 Leiden Observatory, Leiden University,
PO Box 9513,
2300 RA Leiden, The Netherlands
⋆ Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
1
May
2025
Accepted:
2
June
2025
Abstract
Context. Among the most significant chemical functional groups of interstellar molecules are the class of nitriles, which are suggested to be key prebiotic molecules due to their chemical connection to the peptide bond after hydrolysis. The •CN radicals, the simplest representative of this group, have been shown to exhibit strong interactions with interstellar water ices, potentially impacting their reactivity with other radicals nearby.
Aims. This study explores (a) whether CN and •CH3 radicals can readily react to form methyl cyanide (CH3CN) and its isomer methyl isocyanide (CH3NC) and (b) the feasibility of the reaction (CN···H2O)hemi → •C(OH) = NH and its potential role in the formation of acetamide.
Methods. Following a benchmark, we employed density functional theory to map the potential energy surfaces of these chemical processes, focusing on their reactivity on water and carbon monoxide ices.
Results. The results show that CN reacts with •CH3 radicals on water ices, efficiently forming CH3CN and CH3NC. However, these reactions are driven by diffusion of •CH3 towards the reactive site and subsequently compete with back-diffusion of •CH3 from that site. The formation of the radical intermediate •C(OH) = NH on water ice requires quantum tunnelling, and assuming that acetimidic acid forms via CH3 + •C(OH) = NH → CH3C(OH) = NH, it can also only isomerise into acetamide through a sizeable barrier thanks to quantum tunnelling. Both quantum tunnelling-driven reactions are highly dependent on the local structure of the water ice. Finally, radical coupling reactions on carbon monoxide ices are found to be barrierless for all cases, and again both the cyanide and the isocyanide are formed.
Conclusions. This work reinforces the conclusion that •CN radicals on interstellar grain surfaces are highly reactive and unlikely to persist unaltered.
Key words: astrochemistry / molecular processes / solid state: volatile / ISM: molecules
© The Authors 2025
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
The origins of life in the Universe have likely been seeded in the cold and harsh conditions of the interstellar medium (ISM), where chemical evolution occurs alongside star and planet formation Ceccarelli et al. (2023). With over 300 molecular species detected in the ISM, those containing − CN group have been proposed as important prebiotic parent molecules. This is due to the ability of the − CN group to undergo hydrolysation reactions, leading to peptide bonds and amino acids (Goldman et al. 2010), or to be hydrogenated into amines (Raaphorst et al. 2025). Nitrile-bearing molecules comprise a large portion of interstellar molecule detections (∼ 15%), and in particular, simple cyanides such as hydrogen cyanide (HCN) or methyl cyanide CH3CN (also called acetonitrile) are widely observed along different stages of the star- and planet-forming processes.
This study focuses on the formation of methyl cyanide (CH3CN) and its isomer, methyl isocyanide (CH3NC). Methyl cyanide has been detected in the gas phase in a wide range of interstellar environments, including starless and prestellar cores (e.g. Megías et al. 2023; Scibelli et al. 2024), low- and high-mass protostars (e.g. Bergner et al. 2017; Nazari et al. 2021, 2022; Bianchi et al. 2022), protoplanetary disks (e.g. Öberg et al. 2015; Ilee et al. 2021), and Solar System bodies such as comets (e.g. Le Roy et al. 2015; Altwegg et al. 2019). In addition, methyl cyanide has recently been tentatively detected in interstellar ices (Nazari et al. 2024). This interstellar complex organic molecule can form either in the gas phase or on interstellar ices.
Recently, Giani et al. (2023) and Mancini et al. (2024) have revised the main gas-phase formation and evolution pathways of acetonitrile in the gas phase. Giani et al. identified ten reactions contributing to its synthesis, four of which are particularly significant: CH3+ + HCN → CH3CNH+ + hν and CH3OH2+ + HNC → CH3CNH+ + H2O, followed by either dissociative electron recombination or an acid-base reaction with NH3 to convert CH3CNH+ into neutral CH3CN.
In contrast, the dominant non-energetic solid-phase formation mechanisms on ices are thought to involve the radicalradical coupling reactions, and the successive hydrogenation of CCN, (Loomis et al. 2018, reaction 2):
Reactions 1a and 1b (Garrod & Herbst 2006; Garrod et al. 2008),
![Mathematical equation: \[\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{ + CN}} \to \frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{CN,}}\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq1.png) (1a)
(1a)
![Mathematical equation: \[\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{ + CN}} \to \frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{NC,}}\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq2.png) (1b)
(1b)
![Mathematical equation: \[\frame{\text{CCN + H}} \to \cdots \to \frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{CN}}.\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq3.png) (2)
(2)
Interestingly, observational studies of Class 0/I protostars in the Perseus molecular cloud have revealed a correlation between the column densities of methanol and methyl cyanide (Yang et al. 2021). This correlation may point to a shared origin from dust grain surface chemistry or to a chemical connection in the gas phase (Giani et al. 2023; Bianchi et al. 2022).
We focus on the reactions 1a and 1b, building on the findings of CN hydrogenation by Enrique-Romero & Lamberts (2024). That work showed CN hydrogenation leads to HCN and HNC efficiently on H2O and CO ices. On water ice, CN interacts strongly through hemibonding, which nonetheless only minimally hinders reaction with atomic H, yet it imposes a significant activation barrier for reactions with H2. Hemibonding further facilitates the formation of HO•CNH, a precursor to formamide (Rimola et al. 2018). On CO ice, CN interactions are weaker, allowing barrierless hydrogenation with atomic H, while reactions with H2 retain a barrier (∼12 kJ mol−1) similar to the one in the gas phase. •CN can react with CO molecules too, forming NC•CO, which can barrierlessly become hydrogenated into HCOCN by atomic hydrogen.
The goals of this work are to go beyond reactivity with hydrogen and thus investigate (a) how •CN radicals react with •CH3 radicals on interstellar ices to form CH3CN and CH3NC, (b) the role played by diffusion of •CH3 (which is explicitly looked into for the first time), and (c) the reactivity of •CN radicals with water molecules from the ice. We used computational quantum chemical tools as outlined in the methods section (Sect. 2), and the results are presented in Sect. 3. In Sect. 4, we examine the implications of our findings in the context of astrochemistry. And finally, Sect. 5 provides a summary of the main conclusions of this work.
2 Methodology
Density functional theory (DFT) calculations were performed using ORCA 6.0.1 (Neese et al. 2020; Neese 2022). We performed a benchmark study where different reference methods were employed. The explicitly correlated coupled-cluster CCSD(T)-F12 method (Knizia et al. 2009) was used for non-radical-radical reactions and molecule-surface interactions, while the N-electron valence state perturbation theory (NEVPT2) (Angeli et al. 2001, 2007) method was employed for radical-radical reactions, both using Molpro (Werner et al. 2012, 2020). In addition, the complete active space perturbation theory up to second order (CASPT2) method (Werner 1996; Celani & Werner 2000), which is based on the automated construction of atomic valence active space (AVAS) (Sayfutyarova et al. 2017), was also used using both Molpro and OpenMolcas (Li Manni et al. 2023; Battaglia et al. 2023) to double-check our reference values for radical-radical reactions. All calculations employed triple-zeta quality basis sets including diffuse and polarisation functions (see below). For further information on the active space orbitals and raw energetics of the benchmark study, we refer to section Sect. A.
Based on our benchmark, we concluded that the BHandHLYP functional (Becke 1993) combined with the D4 correction (Caldeweyher et al. 2017) for long-range interactions and the def2-TZVPD basis set (Hellweg & Rappoport 2015) provides an appropriate level of theory for modelling the •CH3 + •CN reactivity and •CH3−H2O interactions. There are two cases where BHandHLYP underperforms: the interaction of •CH3 with carbon monoxide (as previously reported by Lamberts et al. 2019) and the •CH3 + •CN → CH3NC reaction. This is not a problem for the former, as we discussed in Sect. 3.3. For the latter, the meta-GGA RSCAN-D3(BJ) functional shows a better performance, and therefore, we used it for this particular reaction.
All DFT calculations encompass geometry optimisations, transition state searches, intrinsic reaction coordinate calculations to connect stationary points along the reaction pathway on the potential energy surface (PES), PES scans, nudged elastic band (NEB) calculations (Ásgeirsson et al. 2021), and frequency calculations to ensure that our geometries correspond to minima or first-order saddle points for transition states. Tight self-consistent field convergence criteria and dense integration grids (DEFGRID3) were adopted throughout this work in ORCA. Unrestricted Kohn-Sham orbitals were used consistently, and for all open-shell singlet calculations, we applied the broken spin symmetry approach using ORCA’s spin-flip routine.
3 Results
In this section, we break down all the results obtained for the reactivity of •CN with •CH3 on H2O and CO ices as well as the reactivity of •CN with a water molecule leading to the intermediate radical HO•CNH, which may subsequently react with •CH3. A summary of all the reactions studied can be found in Table 2.
3.1 Reactivity on water ices
Radical-radical coupling
The principal binding mode of •CN radicals in water is through the formation of hemi-bonded complexes, that is, two-centre and three-electron bonds (Enrique-Romero & Lamberts 2024). Based on the high reactivity of •CN in this binding mode with H atoms (Enrique-Romero & Lamberts 2024), we considered the possibility that it may react with a •CH3 radical instead. In this case, we considered the reactions of C − C and C − N bond formation. As shown in Table 2, both channels sport an activation energy barrier. The mechanism involves breaking the (H2O − •CN)hemi hemibond as the C− C or C− N bonds form. The barriers for C− C and C− N bond formation are 2.8 and 3.1 kJ mol−1 , respectively. We note that the latter was calculated at the RSCAN-D3(BJ) level (see Sect. 2 and Table 1).
In addition to the hemibond, the binding energy distribution of •CN also includes a weaker hydrogen-bonded mode. However, this mode has much less astrochemical significance, as it constitutes only a minimal portion of the binding energy distribution. This interaction leads to a barrierless coupling, consistently proceeding through the C− C bond-forming channel towards CH3CN. Notably, the absence of a barrier implies that the rate-determining step of the reaction is the diffusion of •CH3.
Water-assisted hydrogen transfer
Apart from radical couplings, an alternative reaction pathway involves direct reaction with the water matrix. In this case, the hemibonded complex (H2O− •CN)hemi can evolve into the formamide precursor HO − C• = N − H, mediated by quantum tunnelling, following the mechanism proposed by Rimola et al. through water-assisted hydrogen-transfer (wHt) reactions. This scenario is at play if the hemibonded complex is sufficiently long-lived, for example, if it lies in the ice matrix or is not readily destroyed by competing chemical reactions. Depending on the length of the wHt chain, we find that the activation energy barrier for transforming (H2O− •CN)hemi into HO− •C= NH varies. For hydrogen-bonded H2O chains consisting of two, three, and four water molecules, denoted as wHt(2), wHt(3), and wHt(4)—we refer to Figure 2 for wHt(4), the calculated reaction barriers are 51.6, 34.1, and 20.7 kJ mol−1 , respectively. These processes involve the concerted motion of multiple hydrogen atoms and exhibit relatively low but notable tunnelling crossover temperatures of 136, 71, and 126 K, respectively. This mechanism potentially enables further reactions of the intermediate HO − C• = NH, such as with a nearby radical such as •CH3:
![Mathematical equation: \[\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{ + HO}}\frame{ - ^ \bullet }\frame{\text{C}} = \frame{\text{NH}} \to \frame{\text{HO}} - \frame{\text{C}}\left(\nolbrace \frame{\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}} \norbrace\right)\frame{\text{=NH,~}}\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq4.png) (3)
(3)
![Mathematical equation: \[\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{ + HO}}\frame{ - ^ \bullet }\frame{\text{C=NH}} \to \frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{4}}}\frame{\text{ + HOCN}},\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq5.png) (4)
(4)
![Mathematical equation: \[\frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{3}}}\frame{\text{ + HO}}\frame{ - ^ \bullet }\frame{\text{C=NH}} \to \frame{\text{C}}\frame{\frame{\text{H}}_\frame{\text{4}}}\frame{\text{ + OCNH}}.\]](/articles/aa/full_html/2025/07/aa55360-25/aa55360-25-eq6.png) (5)
(5)
Reaction 3 leads to the formation of acetimidic acid (HO − C(CH3) = NH), a low-barrier product and one of the C2H5NO isomers, along with acetamide and 1-aminoethanol (see Section 4). We found the barriers for reaction 3 proceeding from each wHt(i)-formed HO − C• = NH intermediate to be quite low: 2.1, 1.2, and 3.1 kJ mol−1 . These are comparable to the barriers reported for reactions 1a and 1b, and they primarily involve the reorientation of the •CH3 radical. In our simulations, the radical was initially positioned close to the reactive site, thus eliminating the need to consider local diffusion barriers towards the HO•CNH intermediate (see Figure 2).
On the other hand, reactions 4 and 5 result in the formation of cyanic and isocyanic acids along with methane, respectively, through a direct H transfer (dHa), which is depicted in Figures 2b-ii and 3, respectively. These proceed over barriers of 33.2 and 23.3 kJ mol−1 and are associated with high tunnelling crossover temperatures of 740 and 464 K, suggesting that quantum tunnelling may significantly enhance their reaction rates. Since none of the wHt(i) products displayed a conformation with the − NH group exposed, the reactant geometry shown in Figure 3 was constructed manually, mirroring the configuration in Figure 2b.
Lastly, it is worth noting that in the work of Rimola et al. (2018), a second wHt step was proposed, namely converting HO•CNH into O•CNH2. This transformation requires the −OH group of HO•CNH to be hydrogen-bonded to the −NH group via a bridging water network. Such a configuration is absent in our wHt(4) case, and longer water chains could result in higher barriers, potentially exceeding the 6.4 kJ mol−1 barrier reported by Rimola and colleagues for their intermediate.
Benchmark on interactions and reactivity relevant for this work.
3.2 Diffusion of •CH3 on H2O
All coupling reactions described above share a common characteristic: They are mediated by •CH3 diffusion towards the reactive site, and once there, there is a competition between the reaction and the diffusion of highly mobile •CH3 away from the reaction site. To investigate this behavior, we calculated diffusion barriers for •CH3 on the water ice clusters. We studied the potential effect of having the cyanide radical hemibonded on the surface by testing two scenarios: one with •CN hemibonded to the ice surface (•CN− (H2O)14) and one on the bare (H2O)14 ice. For a more detailed look at the diffusion stationary points and energetics, we refer the reader to Appendix B.1.
We prepared the initial binding site by taking the reactant structure for CH3CN formation (Figure 1a) with transition states identified for •CH3 moving one binding site further from •CN. For the case without •CN on the ice surface, •CH3 started from the same binding site, where the cyanide radical was removed and the ice reoptimised. (See Table 3 and Figure 4 for the energies and examples of the diffusion trajectory.) In both cases, we reproduced the displacement of •CH3 to the nearest four neighbouring sites.
When comparing the BHandHLYP-D4 energies in Table 3, one should take into account that these diffusion pathways are more likely to take place in the exothermic direction. Therefore, while the average barrier height seems to be somewhat lower when moving away from an •CN site (2.3 kJ mol−1) and moving towards a pure water cluster (2.8 kJ mol−1), most pathways leading away from the cyanide are endothermic. In other words, once a methyl radical is close to the cyanide reactant, it is less likely to leave, and we expect back-diffusion to be effectively suppressed.
Including ZPVE corrections only has a small effect, at most 0.8 kJ mol−1 . We caution that the transition states identified in these diffusion calculations are characterised by very low (harmonic) imaginary frequencies, ∼ 10−90 cm−1, and they should therefore be interpreted with care. Additionally, the ZPVE is likely overestimated because long-range Van der Waals interactions are typically anharmonic. Nevertheless, we confirmed that for all cases, the imaginary frequency corresponds to a translational mode of •CH3 parallel to the surface.
Astrochemical models typically approximate diffusion barriers as a fixed fraction of a molecule’s binding energy, commonly by using values between 0.3 and 0.5 for species adsorbed on water ices, despite there being no fundamental physical justification for such a uniform scaling factor choice (Cuppen et al. 2024). In astrochemical databases, typical binding energies for the methyl radical are 9.8 kJ mol−1 (UMIST; McElroy et al. 2013) and 13.3 kJ mol−1 (KIDA; Wakelam et al. 2015). On the other hand, computational studies provide broader estimates for the binding energy of •CH3 on amorphous water ice. For example, Ferrero et al. (2020) report values ranging from 9.1 to 13.3 kJ mol−1 , while Duflot et al. (2021) found a wider span of 7.7−16.1 kJ mol−1 . Applying the standard 0.5 scaling (e.g. as used in UCLCHEM; Holdship et al. 2017) to the lowest reported binding energy (7.7 kJ mol−1) yields a diffusion barrier of approximately 3.8 kJ mol−1 . This value is still higher than the diffusion barriers obtained in this work, suggesting that the commonly used approximation may underestimate the mobility of certain radicals under interstellar conditions.
Summary of the reactions studied in this work, organised according to the binding mode of •CN on either water or carbon monoxide.
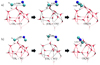 |
Fig. 1 Snapshots of the potential energy surface stationary points for the formation of (a) CH3CN and (b) CH3NC on the (H2O)14 ice cluster, where the •CN radical is initially hemibonded to the ice. Distances are in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is as follows: red for O, blue for N, grey for C, and white for H. |
3.3 Reactivity on carbon monoxide ices
The reactivity on carbon monoxide ice is much simpler, and it is similar to the situation without any explicit surface. On a (CO)13 cluster model (Enrique-Romero & Lamberts 2024), the weakly bound •CN radical on the surface reacts with •CH3 to form either CH3CN or CH3NC barrierlessly, depending on the relative orientation of the radicals upon encounter. This has been confirmed via relaxed PES scans starting where the C− C and C− N bonds are stretched up to 3.0 Å. The only difference lies in the relative stability of each product, where CH3CN is lower in energy than CH3NC.
Additionally, as described in Enrique-Romero & Lamberts (2024), •CN radicals may react with surface CO molecules to form NC•CO radicals with a small activation energy barrier of 2.5 kJ mol−1 . If this intermediate is formed and it is not destroyed by subsequent reactions such as H addition, it can react with nearby radicals, such as •CH3. Indeed, we find that acetyl cyanide (CH3C(O)CN) can be formed barrierlessly, too.
The overestimation arising from the use of BHandHLYP-D4 for the•CH3 interaction to carbon monoxide (see Sect. 2) does not impact the results since a barrierless pathway is identified regardless of the interaction strength. In other words, despite the interaction being predicted to be too strong, this seems not to hamper reactivity. Therefore, we decided not to recalculate the trajectories.
4 Discussion and astrophysical implications
Our results are summarised in Table 2 and Figure 6. The most important result is that for •CN hemibonded to water (its most important binding mode), as both the carbon and nitrogen atoms are available for reaction to form CH3CN and CH3NC with •CH3, respectively. Hence, the hemibond does not impose any chemical restrictions on radical coupling. This aligns with our previous findings for the reactions •CN + •H → HCN and •CN + •H → HNC.
For this radical coupling mechanism, we adopted the common assumption in radical-radical chemistry regarding the availability of free H atoms on the ice. Specifically, this mechanism is expected to be active at intermediate temperatures, where the residence time of H atoms (the primary destroyers of radical species on the ice) is very short. Consequently, the formation of HCN and HNC would have occurred at lower temperatures and preceded the formation of CH3CN and CH3NC.
Even if the abundance of H atoms on the surface is low enough to allow for the reactivity of radicals and CH3 is mobile, the activation energy for the formation of CH3CN and CH3NC is as low as the diffusion barriers for CH3. This is because these barriers stem from the break of the weak interactions of mainly CH3 with the surface to move into the same binding site as CN. Consequently, the competition between product formation and back diffusion plays a crucial role in these reactions, which could affect the final efficiency of these reaction paths. This of course also applies to the chemistry on CO ice, where the interactions of CH3 and CN are even weaker. In any case, we note that the radical coupling reactions under discussion are not the only pathway for the formation of methyl cyanide and isocyanide. For example, on surfaces, reaction 2 also plays a role, while additional pathways exist in the gas phase (see Sect. 1). Determining the relative importance of these pathways a priori is challenging, as it requires dedicated study of the reaction 2 and astrochemical modelling. These two aspects will be the focus of a forthcoming work, and we defer this discussion to a later stage.
Regardless of its formation mechanism, methyl cyanide is a parent species for other molecules with high prebiotic interest in the ISM. One such molecule is acetamide (Am; CH3C(O)NH2) , first detected nearly two decades ago in the star-forming region Sagittarius B2(N) (Hollis et al. 2006), where it is abundant. It is comparable to formamide, with which it is consistently correlated, and acetaldehyde (Halfen et al. 2011). Notably, acetamide is one of the largest interstellar molecules detected with a peptide bond − so far the largest is glycolamide by Rivilla et al. (2023). More recently, acetamide has been observed in various protostellar environments (e.g. Colzi et al. 2021; Ligterink et al. 2022; Zeng et al. 2023) as well as on comet 67P/Churyumov−Gerasimenko by the Rosetta mission (Goesmann et al. 2015; Altwegg et al. 2017). Several formation pathways for acetamide have been proposed, including gas-phase ion-molecule reactions (Quan & Herbst 2007; Halfen et al. 2011; Redondo et al. 2014), ice-surface processing (e.g. Frigge et al. 2018; Ligterink et al. 2018; Drabkin et al. 2023), and hydrogen abstraction from formamide (HCONH2), followed by reaction with •CH3 (Belloche et al. 2017). We propose an alternative path to form acetamide. As shown by Rimola et al. (2018), hemibonded •CN can evolve into OH− •C= NH via a water-assisted H-transfer mechanism. We reproduced this mechanism, showing its dependence on the length of the H transfer relay length of the water ice, which in turn also affects the isomery of the radical (cis or trans-HO•CNH). Depending on the water chain length and the method of choice, barriers ranging between 16.1 and 70.6 kJ/mol have been found, including in the works by Rimola et al. (2018) and Silva-Vera et al. (2024), and we argue that the lower barriers can be overcome thanks to quantum tunnelling. Once OH− •C= NH is formed, Rimola et al. (2018) proposes a follow-up water-assisted H-transfer reaction (with a local barrier of 6.4 kJ mol−1), which should again somewhat depend on the local properties of the ice structure. Instead, we propose that once OH− •C= NH is formed, it may react with a nearby radical, in this work a •CH3 radical, forming acetimidic acid (AAc; CH3C(OH)NH), a higher energy isomer of acetamide. For the three cases presented, the barrier of this follow-up reaction is of the same order as the barriers for the diffusion of CH3, between 1.2 and 3.1 kJ mol−1 (see Table 2, and the SI for more information).
Finally, an alternative reaction path involves the direct H transfer from the intermediate OH− •C= NH radical by •CH3. Such a reaction would lead to either cyanic acid (HOCN) or isocyanic acid (HNCO; see Figures 2b and 3). The barriers for each of these channels are 23.3 and 33.2 kJ mol−1 , respectively, indicating that the coupling reaction leading to CH3C(OH)NH) is far more efficient. For the sake of comparison, and given the potential importance of cyanic and isocyanic acid on interstellar ices, whose conjugate base is OCN− commonly detected in interstellar ice IR adsorption bands (e.g. Boogert et al. 2015; McClure et al. 2023), we have also studied these reactions sub-stituting •CH3 by an H atom, which results in the lowering of the barriers down to 6.0 and 23.2 kJ mol−1 for the formation of cyanic and isocyanic acid, respectively (see Figure C.1 in the appendix). The barriers for each reaction keep the same relative ordering as for the reactions with the methyl radical. Still, this mechanism would have to compete with the H addition leading to HOCHNH (hydroxymethylimine). Therefore, these seem to be rather inefficient ways to form cyanic and isocyanic acid. Coming back to acetamide, our proposed mechanism aligns with the pathway suggested by Belloche et al. (2017), linking acetamide and formamide, and it also incorporates elements of the ice-surface processing route described by Drabkin et al. (2023); namely, we propose acetimidic acid as a precursor of acetamide. Once acetimidic acid is formed, it can undergo ketoenol tautomerism facilitated by the ice surface, converting it into acetamide through a water-assisted H-transfer reaction. To explore this, we manually prepared two binding modes shown in Figures 5a and b in a way that there is a clear H-transfer relay path. The differences between the two modes are the interaction of AAc with the cluster and the number of water molecules participating in the isomerisation. The first binding mode exhibits a high activation barrier of 58.1 kJ mol−1 and a low crossover temperature of 58 K, with a reaction occurring over four water molecules. In contrast, the second binding mode presents a significantly lower barrier of just 20.0 kJ mol−1 , which is comparable to the lowest energy barrier for the (•CN− H2O)hemi → HO•CNH conversion; has a crossover temperature of 134 K; and requires only two water molecules. This highlights the critical role of the binding mode and water’s catalytic influence in facilitating H-transfer reactions and the strong impact of the local ice structure on reaction energetics.
On the other hand, on CO ices, the picture is much simpler. The reaction of •CH3 with either •CN or NC•CO (the product of cyanide’s chemisorption on CO ice) is barrierless, leading to CH3CN, CH3NC, or CH3C(O)CN (acetyl cyanide). In these cases, competition between diffusion and reaction is also expected, especially given the much-reduced binding energies on CO ices. For example, •CH3 has a binding energy of 1.7 kJ mol−1 (∼ 200 K) on CO (Lamberts et al. 2019), while it takes values between 8.3 and 13.3 kJ mol−1 (∼1000−1600 K) on amorphous solid water Ferrero et al. (2020); Bovolenta et al. (2022).
Finally, we highlight that despite the technical challenges associated with working with nitriles (e.g. HCN) in the laboratory (mainly due to their toxicity) and the inherent difficulties of handling radical species, exploring these reaction pathways experimentally would provide valuable insights into the final products of the overall reaction network. To date, no dedicated experimental studies have addressed the formation of CH3CN and CH3NC on interstellar ice analogues other than those focused on the energetic processing of ices (e.g. Volosatova et al. 2021; Canta et al. 2023; Chuang et al. 2024). Regardless, the formation of CH3CN on interstellar ices remains a particularly intriguing subject, as this species is believed to serve as a precursor to more complex molecules, such as amino acids, amines, and amides (e.g. Hudson et al. 2008; Danger et al. 2011; Bulak et al. 2021). As such, further experimental investigation is certainly warranted.
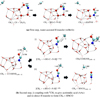 |
Fig. 2 Snapshots of the potential energy surface stationary points for (a) the (H2O− •CN)hemi → HO•C= NH reaction (wHt(4)) and (b) a second step where acetimidic acid is formed via •CH3 + HO•C= NH → CH3C(OH)NH (i) and an alternative path leading to CH4 + HNCO through a direct H-transfer reaction. Distances are provided in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
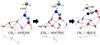 |
Fig. 3 Snapshots of the potential energy surface stationary points to form cyanic acid (HOCN) from the direct H-abstraction reaction HO•CNH + CH3 on the (H2O)14 cluster. The reactant’s geometry was prepared manually to mimic that in Fig. 2b. Distances are in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
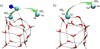 |
Fig. 4 Diffusion of •CH3 from the initial binding site (BS0) to the neighbouring binding site BS3 for two scenarios: a) •CN− (H2O)14 and the bare ice b) (H2O)14. The arrows indicate the motion the CH3 radical follows when it hops to BS3. The initial BS0 geometry in the left panel corresponds to the reactants’ geometry for the formation of CH3CN and CH3NC in Figure 1. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
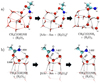 |
Fig. 5 Snapshots of the potential energy surface stationary points for the acetimidic acid to acetamide isomerisation. Panel a corresponds to the high barrier isomerisation reaction case, while the panel b corresponds to the lower barrier one. Distances are provided in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
 |
Fig. 6 Summary of the reactions studied in this work between •CH3 + •CN on water and carbon monoxide ice surfaces. The first line indicates the possible outcomes of diffusive chemistry, while the second indicates the products after the reaction with one of the ice molecules from the surface. We note that after chemisorption, reaction products can proceed from two different mechanisms: chemisorption followed by H-abstraction (cyanic and isocyanic acid) or chemisorption followed by radical-radical coupling (acetimidic acid and acetyl cyanide). |
5 Conclusions
This work further supports the idea that depleted •CN radicals on interstellar dust grains cannot be ‘preserved’ for future chemical processes if they remain on the surface. In Enrique-Romero & Lamberts (2024), we demonstrated that •CN is readily converted into HCN or HNC, except in environments with insufficient atomic H. Here, we have shown that •CN is also highly reactive with fast-diffusing radicals such as •CH3, as •CN itself is expected to be largely immobile due to its unusually strong hemibonded interaction with water ice surfaces. The reaction •CH3 + •CN leads to the formation of both methyl cyanide (CH3CN) and methyl isocyanide (CH3NC), and these reactions compete against •CH3 back-diffusion. Additionally, even if •CN does not react to form HCN, HNC, CH3CN, or CH3NC on water ices, it can still react with a water molecule to form HO•CNH, as shown earlier by Rimola et al. (2018). This newly formed radical is related to formamide (HCONH2) via a reaction with H, acetimidic acid (CH3COHNH) via reaction with CH3, and acetamide (CH3CONH2) following a water-assisted isomerisation.
On the other hand, on CO ices, both CH3CN and CH3NC form without an energy barrier. Likewise, CH3C(O)CN can form when •CN reacts with surface CO, leading to NC•CO. As with previous cases, these reactions compete with radical diffusion.
Finally, we point out that there are more surface routes to form methyl cyanide, such as •CCN hydrogenation, which will be the focus of a forthcoming work. Therefore, further experimental studies on these reaction paths would certainly be of interest to the community.
Data availability
Extra supporting information can be found in the Zenodo repository with DOI: 10.5281/zenodo.15630762 (link).
Acknowledgement
We thank Dr. G. Molpeceres for stimulating discussions, especially regarding the AVAS calculations in MOLPRO. This project has received funding from the Horizon Europe Framework Programme (HORIZON) under the Marie Skłodowska-Curie grant agreement No 101149067, “ICE-CN”. Finally, this work was granted access to the HPC resources of the high-performance computer SNELLIUS, part of the SURF cooperative of educational and research institutions in The Netherlands, under project No EINF-6197.
Appendix A Benchmark
This section presents the structures used in our benchmark study; corresponding energetic data are available in the Zenodo repository (link). For multireference calculations, active space selection varied by software. In MOLPRO, we used an automated method to include relevant atomic orbitals: the 2pz of C in CH3, the 2pi of O in water aligned with CN hemibonding, and the full valence of CN (alignment ensured proper selection). A figure showing the resulting orbitals before and after the CASPT2 step is available from the online repository, named as Figure A3. In OPENMOLCAS, full-valence selection was limited by memory, so we manually defined an active space of 24 orbitals and 15 electrons from canonical UHF orbitals, focusing on orbitals centred on reacting atoms.
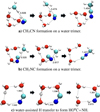 |
Fig. A.1 Structures used in the reactivity benchmark. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. |
 |
Fig. A.2 Structures used in the interactions benchmark. From left to right: Cases 1 and 2 on the water trimer, and cases 1 and 2 on a single CO molecule. Atomic colouring code: white for hydrogen, grey for carbon, and red for oxygen. |
Appendix B Methyl radical diffusion
Figure B.1 shows the binding sites used in the diffusion study. Energetic and transition state data are provided in Table B1 in the Zenodo repository (link).
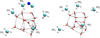 |
Fig. B.1 Binding sites explored to study the diffusion of •CH3 on water with (left panel) and without (right panel) CN on the surface. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. |
Appendix C Figures for HO•CNH + •H → HOCN/HNCO + H2
We have studied the H-abstraction by •H from HO•CNH by just taking the structures for the reactions with •CH3 and substituting the methyl radical by the hydrogen atom, then finding the stationary points of the reaction, as shown in Figure C.1.
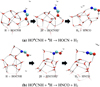 |
Fig. C.1 Steps of the H-abstraction reactions to form cyanic and isocyanic acid from HO•CNH + •H. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. The energetics can be found in the repository (link), in Table D4. |
Appendix D Reactivity with molecules from the ice
Tables D1−D4 in the Zenodo repository (link) report activation and reaction energies. Table D1 covers radical−radical couplings to form CH3CN and CH3NC, while Tables D2−D4 include water-assisted H transfers and their follow-up reactions: D2 (wHt over 2−3 waters), D3 (wHt over 4 waters + abstraction by •CH3), and D4 (abstraction by H atoms).
References
- Altwegg, K., Balsiger, H., Berthelier, J. J., et al. 2017, MNRAS, 469, S130 [Google Scholar]
- Altwegg, K., Balsiger, H., & Fuselier, S. A. 2019, ARA&A, 57, 113 [NASA ADS] [CrossRef] [Google Scholar]
- Angeli, C., Cimiraglia, R., Evangelisti, S., Leininger, T., & Malrieu, J.-P. 2001, JCP, 114, 10252 [Google Scholar]
- Angeli, C., Pastore, M., & Cimiraglia, R. 2007, Theor. Chem. Acc., 117, 743 [Google Scholar]
- Ásgeirsson, V., Birgisson, B. O., Bjornsson, R., et al. 2021, J. Chem. Theory Comput., 17, 4929 [CrossRef] [Google Scholar]
- Battaglia, S., Galván, I. F., & Lindh, R. 2023, in Theoretical and Computational Photochemistry (Amsterdam: Elsevier), 135 [Google Scholar]
- Becke, A. D. 1993, JCP, 98, 5648 [Google Scholar]
- Belloche, A., Meshcheryakov, A. A., Garrod, R. T., et al. 2017, A&A, 601, A49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bergner, J. B., Öberg, K. I., Garrod, R. T., & Graninger, D. M. 2017, ApJ, 841, 120 [Google Scholar]
- Bianchi, E., Ceccarelli, C., Codella, C., et al. 2022, A&A, 662, A103 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Boogert, A. C. A., Gerakines, P. A., & Whittet, D. C. B. 2015, ARA&A, 53, 541 [Google Scholar]
- Bovolenta, G. M., Vogt-Geisse, S., Bovino, S., & Grassi, T. 2022, ApJS, 262, 17 [NASA ADS] [CrossRef] [Google Scholar]
- Bulak, M., Paardekooper, D. M., Fedoseev, G., & Linnartz, H. 2021, A&A, 647, A82 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Caldeweyher, E., Bannwarth, C., & Grimme, S. 2017, JCP, 147, 034112 [Google Scholar]
- Canta, A., Öberg, K. I., & Rajappan, M. 2023, ApJ, 953, 81 [NASA ADS] [CrossRef] [Google Scholar]
- Ceccarelli, C., Codella, C., Balucani, N., et al. 2023, ASP Conf. Ser., 534, 379 [NASA ADS] [Google Scholar]
- Celani, P., & Werner, H.-J. 2000, JCP, 112, 5546 [Google Scholar]
- Chuang, K. J., Jäger, C., Santos, J. C., & Henning, T. 2024, A&A, 687, A7 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Colzi, L., Rivilla, V. M., Beltrán, M. T., et al. 2021, A&A, 653, A129 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cuppen, H. M., Linnartz, H., & Ioppolo, S. 2024, ARA&A, 62, 243 [Google Scholar]
- Danger, G., Bossa, J. B., de Marcellus, P., et al. 2011, A&A, 525, A30 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Drabkin, V. D., Volosatova, A. D., & Feldman, V. I. 2023, MNRAS, 518, 1744 [Google Scholar]
- Duflot, D., Toubin, C., & Monnerville, M. 2021, Front. Astron. Space Sci., 8, 24 [NASA ADS] [CrossRef] [Google Scholar]
- Enrique-Romero, J., & Lamberts, T. 2024, JCPL, 15, 7799 [Google Scholar]
- Ferrero, S., Zamirri, L., Ceccarelli, C., et al. 2020, ApJ, 904, 11 [NASA ADS] [CrossRef] [Google Scholar]
- Frigge, R., Zhu, C., Turner, A. M., et al. 2018, ApJ, 862, 84 [NASA ADS] [CrossRef] [Google Scholar]
- Garrod, R. T., & Herbst, E. 2006, A&A, 457, 927 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Garrod, R. T., Widicus Weaver, S. L., & Herbst, E. 2008, ApJ, 682, 283 [Google Scholar]
- Giani, L., Ceccarelli, C., Mancini, L., et al. 2023, MNRAS, 526, 4535 [NASA ADS] [CrossRef] [Google Scholar]
- Goesmann, F., Rosenbauer, H., Bredehöft, J. H., et al. 2015, Science, 349, 2.689 [CrossRef] [Google Scholar]
- Goldman, N., Reed, E. J., Fried, L. E., William Kuo, I. F., & Maiti, A. 2010, Nat. Chem., 2, 949 [NASA ADS] [CrossRef] [Google Scholar]
- Halfen, D. T., Ilyushin, V., & Ziurys, L. M. 2011, ApJ, 743, 60 [Google Scholar]
- Hellweg, A., & Rappoport, D. 2015, PCCP, 17, 1010 [Google Scholar]
- Holdship, J., Viti, S., Jiménez-Serra, I., Makrymallis, A., & Priestley, F. 2017, ApJ, 154, 38 [CrossRef] [Google Scholar]
- Hollis, J., Lovas, F. J., Remijan, A. J., et al. 2006, ApJ, 643, L25 [NASA ADS] [CrossRef] [Google Scholar]
- Hudson, R. L., Moore, M. H., Dworkin, J. P., Martin, M. P., & Pozun, Z. D. 2008, Astrobiology, 8, 771 [Google Scholar]
- Ilee, J. D., Walsh, C., Booth, A. S., et al. 2021, ApJS, 257, 9 [NASA ADS] [CrossRef] [Google Scholar]
- Knizia, G., Adler, T. B., & Werner, H.-J. 2009, JCP, 130, 054104 [Google Scholar]
- Lamberts, T., Markmeyer, M. N., Kolb, F. J., & Kästner, J. 2019, ACS Earth Space Chem., 3, 958 [Google Scholar]
- Le Roy, L., Altwegg, K., Balsiger, H., et al. 2015, A&A, 583, A1 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Li Manni, G. Fdez. Galva’an, I., Alavi, A., et al. 2023, J. Chem. Theory Comput., 19, 6933 [Google Scholar]
- Ligterink, N. F. W., Terwisscha van Scheltinga, J., Taquet, V., et al. 2018, MNRAS, 480, 3628 [Google Scholar]
- Ligterink, N. F. W., Ahmadi, A., Luitel, B., et al. 2022, ACS Earth Space Chem., 6, 455 [NASA ADS] [CrossRef] [Google Scholar]
- Loomis, R. A., Cleeves, L. I., Öberg, K. I., et al. 2018, ApJ, 859, 131 [Google Scholar]
- Mancini, L., Valença Ferreira de Aragão, E., Pirani, F., et al. 2024, A&A, 691, A83 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- McClure, M. K., Rocha, W. R. M., Pontoppidan, K. M., et al. 2023, Nat. Astron., 7, 431 [NASA ADS] [CrossRef] [Google Scholar]
- McElroy, D., Walsh, C., Markwick, A. J., et al. 2013, A&A, 550, A36 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Megías, A., Jiménez-Serra, I., Martín-Pintado, J., et al. 2023, MNRAS, 519, 1601 [Google Scholar]
- Nazari, P., van Gelder, M. L., van Dishoeck, E. F., et al. 2021, A&A, 650, A150 [EDP Sciences] [Google Scholar]
- Nazari, P., Meijerhof, J. D., van Gelder, M. L., et al. 2022, A&A, 668, A109 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Nazari, P., Rocha, W. R. M., Rubinstein, A. E., et al. 2024, A&A, 686, A71 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Neese, F. 2022, WIREs Comput. Mol. Sci., 12, 5 [Google Scholar]
- Neese, F., Wennmohs, F., Becker, U., & Riplinger, C. 2020, JCP, 152, 224108 [Google Scholar]
- Öberg, K. I., Guzmán, V. V., Furuya, K., et al. 2015, Nature, 520, 198 [Google Scholar]
- Quan, D., & Herbst, E. 2007, A&A, 474, 521 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Raaphorst, M. T., Enrique-Romero, J., & Lamberts, T. 2025, ACS Earth Space Chem., 9, 1534 [Google Scholar]
- Redondo, P., Barrientos, C., & Largo, A. 2014, ApJ, 793, 32 [NASA ADS] [CrossRef] [Google Scholar]
- Rimola, A., Skouteris, D., Balucani, N., et al. 2018, ACS Earth Space Chem., 2, 720 [Google Scholar]
- Rivilla, V. M., Sanz-Novo, M., Jiménez-Serra, I., et al. 2023, ApJ, 953, L20 [NASA ADS] [CrossRef] [Google Scholar]
- Sayfutyarova, E. R., Sun, Q., Chan, G. K.-L., & Knizia, G. 2017, J. Chem. Theory Comput., 13, 4063 [Google Scholar]
- Scibelli, S., Shirley, Y., Megías, A., & Jiménez-Serra, I. 2024, MNRAS, 533, 4104 [Google Scholar]
- Silva-Vera, G., Bovolenta, G. M., Rani, N., Vera, S., & Vogt-Geisse, S. 2024, ACS Earth Space Chem., 8, 1480 [Google Scholar]
- Volosatova, A. D., Lukianova, M. A., Zasimov, P. V., & Feldman, V. I. 2021, PCCP, 23, 18449 [Google Scholar]
- Wakelam, V., Loison, J. C., Herbst, E., et al. 2015, ApJS, 217, 20 [NASA ADS] [CrossRef] [Google Scholar]
- Werner, H.-J. 1996, Mol. Phys., 89, 645 [NASA ADS] [CrossRef] [Google Scholar]
- Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., & Schütz, M. 2012, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2, 242 [CrossRef] [Google Scholar]
- Werner, H.-J., Knowles, P. J., Manby, F. R., et al. 2020, JCP, 152, 144107 [Google Scholar]
- Yang, Y.-L., Sakai, N., Zhang, Y., et al. 2021, ApJ, 910, 20 [Google Scholar]
- Zeng, S., Rivilla, V. M., Jiménez-Serra, I., et al. 2023, MNRAS, 523, 1448 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
Summary of the reactions studied in this work, organised according to the binding mode of •CN on either water or carbon monoxide.
All Figures
|
Fig. 1 Snapshots of the potential energy surface stationary points for the formation of (a) CH3CN and (b) CH3NC on the (H2O)14 ice cluster, where the •CN radical is initially hemibonded to the ice. Distances are in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is as follows: red for O, blue for N, grey for C, and white for H. |
| In the text | |
|
Fig. 2 Snapshots of the potential energy surface stationary points for (a) the (H2O− •CN)hemi → HO•C= NH reaction (wHt(4)) and (b) a second step where acetimidic acid is formed via •CH3 + HO•C= NH → CH3C(OH)NH (i) and an alternative path leading to CH4 + HNCO through a direct H-transfer reaction. Distances are provided in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
| In the text | |
|
Fig. 3 Snapshots of the potential energy surface stationary points to form cyanic acid (HOCN) from the direct H-abstraction reaction HO•CNH + CH3 on the (H2O)14 cluster. The reactant’s geometry was prepared manually to mimic that in Fig. 2b. Distances are in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
| In the text | |
|
Fig. 4 Diffusion of •CH3 from the initial binding site (BS0) to the neighbouring binding site BS3 for two scenarios: a) •CN− (H2O)14 and the bare ice b) (H2O)14. The arrows indicate the motion the CH3 radical follows when it hops to BS3. The initial BS0 geometry in the left panel corresponds to the reactants’ geometry for the formation of CH3CN and CH3NC in Figure 1. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
| In the text | |
|
Fig. 5 Snapshots of the potential energy surface stationary points for the acetimidic acid to acetamide isomerisation. Panel a corresponds to the high barrier isomerisation reaction case, while the panel b corresponds to the lower barrier one. Distances are provided in Å. Atoms relevant to the reaction are highlighted using a ball-and-stick representation. The atom colour-code is red for O, blue for N, grey for C, and white for H. |
| In the text | |
|
Fig. 6 Summary of the reactions studied in this work between •CH3 + •CN on water and carbon monoxide ice surfaces. The first line indicates the possible outcomes of diffusive chemistry, while the second indicates the products after the reaction with one of the ice molecules from the surface. We note that after chemisorption, reaction products can proceed from two different mechanisms: chemisorption followed by H-abstraction (cyanic and isocyanic acid) or chemisorption followed by radical-radical coupling (acetimidic acid and acetyl cyanide). |
| In the text | |
|
Fig. A.1 Structures used in the reactivity benchmark. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. |
| In the text | |
|
Fig. A.2 Structures used in the interactions benchmark. From left to right: Cases 1 and 2 on the water trimer, and cases 1 and 2 on a single CO molecule. Atomic colouring code: white for hydrogen, grey for carbon, and red for oxygen. |
| In the text | |
|
Fig. B.1 Binding sites explored to study the diffusion of •CH3 on water with (left panel) and without (right panel) CN on the surface. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. |
| In the text | |
|
Fig. C.1 Steps of the H-abstraction reactions to form cyanic and isocyanic acid from HO•CNH + •H. Atomic colouring code: white for hydrogen, grey for carbon, blue for nitrogen, and red for oxygen. The energetics can be found in the repository (link), in Table D4. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.