| Issue |
A&A
Volume 657, January 2022
|
|
|---|---|---|
| Article Number | A85 | |
| Number of page(s) | 6 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202142206 | |
| Published online | 17 January 2022 | |
Ultraviolet and vacuum ultraviolet photo-processing of protonated benzonitrile (C6H5CNH+)
A plausible pathway to larger interstellar aromatics
1
Université Paris-Saclay, CNRS, Institut des Sciences Moléculaires d’Orsay,
91405
Orsay,
France
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2
CNRS, Aix-Marseille Université, Laboratoire PIIM,
Marseille,
France
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
3
Synchrotron SOLEIL, L’Orme des Merisiers,
91192
Saint Aubin,
Gif-sur-Yvette,
France
4
INRAE, UAR1008, Transform Department, Rue de la Géraudière,
BP 71627,
44316
Nantes,
France
5
School of Chemistry, University of New South Wales,
Sydney,
NSW
2052,
Australia
6
Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong,
Wollongong,
New South Wales
2522,
Australia
Received:
13
September
2021
Accepted:
24
November
2021
Abstract
Context. The recent detection in pre-stellar sources of cyano-substituted and pure hydrocarbon cycles has emphasized the importance of aromatic chemistry in the earliest stages of star formation. Ultraviolet (UV) and vacuum-UV (VUV) radiation is ubiquitous in space and thus the photo-processing of small cyclic ions may open a window onto rich chemical networks and lead to the formation of larger aromatics in space.
Aims. The aim is to investigate the fate of protonated benzonitrile species after UV and VUV photoexcitation and the subsequent potential impact on stellar and interstellar chemistry.
Methods. Protonated benzonitrile was isolated in a linear ion trap prior to irradiation with UV and VUV radiation (4.5–13.6 eV) from the DESIRS beamline at synchrotron SOLEIL. The study was extended down to 3.5 eV using a cryogenic Paul ion trap coupled to an OPO laser at the PIIM laboratory. Photodissociation action spectra were obtained by monitoring the photofragment yields as a function of photon energy.
Results. The UV/VUV photodissociation action spectra of protonated benzonitrile show structured bands from 3.8 to 9 eV. The primary dissociation channel of protonated benzonitrile corresponds to HCN/HNC loss and formation of the phenylium cation (C6H5+); whereas at high energies, a minor channel is observed that correlates with HC3N loss and formation of C4H5+.
Conclusions. The UV and VUV photodestruction of protonated benzonitrile leads to the formation of a highly reactive cationic species, C6H5+, predicted to be an important precursor of larger aromatic molecules in space, such as polycyclic aromatic hydrocarbons. The inclusion of C6H5+ – a precursor of benzene and, by extension, of benzonitrile – as the result of formation via the photodissociation of protonated benzonitrile in current astrochemical models could improve the predicted abundance of benzonitrile, which is currently underestimated.
Key words: ultraviolet: ISM / ISM: molecules / techniques: spectroscopic / molecular processes
© U. Jacovella et al. 2022
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
Aromatic molecules are prevalent in the chemistry occurring on Earth, with ~80% of the 135 million compounds registered in Chemical Abstract Service containing at least one five- or six-membered ring (Lipkus et al. 2008). The first detection of such a molecule in space was in 2001, when benzene was identified in absorption towards the proto-planetary nebula CRL 618 by Cernicharo et al. (2001) through its infrared spectral signature. Almost two decades later, McGuire et al. (2018) detected benzonitrile molecules in emission from the dense molecular cloud TMC-1 via radio astronomy and so, the second six-membered ring was found in the interstellar medium (ISM). The elusiveness of aromatic species in space has tantalized astrochemists for decades, especially since the unidentified infrared emission bands (UIBs), which are ubiquitous spectral signatures in a broad variety of astronomical objects, are widely assumed to result from a collective emission of polycyclic aromatic hydrocarbon (PAH) molecules (Allamandola et al. 1989; Schlemmer et al. 1994). For example, among the different products formed after photochemical dissociation of nitrogen and methane (major components of Titan’s atmosphere), benzene has been observed in Titan’s upper atmosphere (Coustenis et al. 2003). Moreover, it is suspected that benzene molecules are at the origin of the organic haze layer that masks Titan’s surface by initiating the formation of larger PAHs or those containing nitrogen, known as PANHs (Delitsky & McKay 2010). Most molecules detected in the ISM possessing an aromatic motif are cyano-substituted species. Benzonitrile was the first to be detected (McGuire et al. 2018; Burkhardt et al. 2021b), followed by 1- and 2-cyano-1 and 3-cyclopentadiene (McCarthy et al. 2021; Lee et al. 2021), then the first polycyclics: 1- and 2-cyanonaphthalene (McGuire et al. 2021). Although it might be surprising that cyano-substituted aromatics were detected before non-substituted aromatic hydrocarbons, this is merely a consequence of the detection tools employed. With the exception of benzene, all these species were detected through their rotational spectra using radio telescopes; as such, a permanent dipole moment is a prerequisite unfulfilled by apolar PAHs. Nevertheless, thanks to the milliKelvin level of sensitivity available with the most modern radio telescopes, Cernicharo et al. (2021b) and Burkhardt et al. (2021a) recently detected the first pure hydrocarbon cycles, ethynyl cyclopropenylidene, cyclopentadiene, and indene in the TMC-1 cloud. In the same way, cyclopropenylidene was recently detected with the Atacama Large Millimeter Array on Titan (Nixon et al. 2020), exhibiting a very complex chemistry. Moreover, benzonitrile is important for Titan’s atmosphere since it has been detected in the tholin pyrolyzates obtained as a minor component in spark-discharge synthesis experiments by Khare et al. (1981). However, the predicted benzonitrile production in the lower atmosphere is weak since CN⋅ is significantly consumed through reaction with CH4 (Loison et al. 2019). While the radical–radical reaction of CN⋅ with the phenyl radical (C6 H ) can also yield benzonitrile Lee et al. (2019), the low abundance predicted by Loison et al. (2019), between 1 × 10−9 at 1000 km to 10−14 at 350 km, will make its detection difficult in Titan’s atmosphere despite the strong dipole moment (4.71 D) that had allowed for its detectionin TMC-1 by McGuire et al. (2018).
) can also yield benzonitrile Lee et al. (2019), the low abundance predicted by Loison et al. (2019), between 1 × 10−9 at 1000 km to 10−14 at 350 km, will make its detection difficult in Titan’s atmosphere despite the strong dipole moment (4.71 D) that had allowed for its detectionin TMC-1 by McGuire et al. (2018).
The formation mechanisms of benzonitrile in pre-stellar (and possibly protostellar sources, where it has been detected) remain elusive (Burkhardt et al. 2021b). Models that satisfactorily reproduce the abundances of carbon chains hugely underpredict the benzonitrile column density compared to the measured value, indicating that interstellar chemistry leading to planar structures is poorly understood. Once formed, benzonitrile itself can be seen as a building block of interstellar aromatics. Indeed, the major hindrance to PAH formation is usually the formation of the first aromatic ring (Cherchneff et al. 1992). The low temperatures of interstellar gas clouds and circumstellar envelopes imply that reactions with energetic barriers are disfavored, whereas barrierless reactions, such as ion-neutral reactions, are predominant pathways in interstellar chemistry.
Following UV/VUV absorption, interstellar molecules can undergo several chemical processes such as photoionization and photodissociation. Cations account for roughly 10% of the known interstellar and circumstellar molecular species. Furthermore, the only conclusively identified carrier of diffuse interstellar bands (DIBs) is a cationic species, C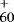 (Campbell et al. 2015). A large portion of these charged species (roughly 80%) are protonated species (Etim et al. 2017). Proton transfer reactions are the most likely routes to form protonated benzonitrile molecules in the ISM. The proton originates typically from H
(Campbell et al. 2015). A large portion of these charged species (roughly 80%) are protonated species (Etim et al. 2017). Proton transfer reactions are the most likely routes to form protonated benzonitrile molecules in the ISM. The proton originates typically from H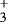 molecules, a strong proton donor (Larsson et al. 2012; Etim et al. 2017). In dense molecular clouds, the primary ionization source is cosmic rays, which subsequently form H
molecules, a strong proton donor (Larsson et al. 2012; Etim et al. 2017). In dense molecular clouds, the primary ionization source is cosmic rays, which subsequently form H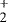 that rapidly transforms into H
that rapidly transforms into H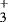 . The large abundance of H
. The large abundance of H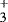 (Geballe & Oka 1996; McCall et al. 1999) provides a substantial reservoir of protons to form protonated molecules. These proton transfer reactions can also occur in photon dominated region’s (PDRs), where H
(Geballe & Oka 1996; McCall et al. 1999) provides a substantial reservoir of protons to form protonated molecules. These proton transfer reactions can also occur in photon dominated region’s (PDRs), where H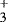 has also been detected in considerable quantities (McCall et al. 1998; Geballe et al. 1999). Despite the infrared signatures of protonated benzonitrile having been recorded in the laboratory (Chatterjee & Dopfer 2018), it has not been detected in the ISM thus far.
has also been detected in considerable quantities (McCall et al. 1998; Geballe et al. 1999). Despite the infrared signatures of protonated benzonitrile having been recorded in the laboratory (Chatterjee & Dopfer 2018), it has not been detected in the ISM thus far.
Ultraviolet photons are not merely an important source of ionization but they also drive important chemical processes initiated by photodissociation. Indeed, UV photons are efficient at destroying molecular ions, especially in environments exposed to strong UV fluxes, such as PDRs, circumstellar envelopes, and circumstellar disks. Nevertheless, experimental data on the photolysis of medium to large ions is still missing despite being crucial to correctly model chemical evolution in the ISM. The photodissociation of protonated benzonitrile (BNH+) is expected to lead to the formation of phenylium cation (C6 H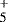 ) (Freiser & Beauchamp 1977; Wincel et al. 1990). C6 H
) (Freiser & Beauchamp 1977; Wincel et al. 1990). C6 H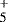 is highly reactive and thus must play a significant role in the ion-neutral chemical networks in space (Ascenzi et al. 2007; Soliman et al. 2012). The recent detection of neutral benzonitrile in different pre-stellar sources emphasized the importance of aromatic chemistry at the earliest stages of star formation. While the presence and the role of protonated benzonitrile species remains to be determined, photodissociation of BNH+ could open up unexplored chemical networks.
is highly reactive and thus must play a significant role in the ion-neutral chemical networks in space (Ascenzi et al. 2007; Soliman et al. 2012). The recent detection of neutral benzonitrile in different pre-stellar sources emphasized the importance of aromatic chemistry at the earliest stages of star formation. While the presence and the role of protonated benzonitrile species remains to be determined, photodissociation of BNH+ could open up unexplored chemical networks.
The present work investigates the fate of BNH+ after UV/VUV photoexcitation from 3.5 to 13.6 eV and its potential importance in the bottom up formation of PAHs in the ISM. The results presented in the article have been obtained using VUV radiation from the SOLEIL synchrotron coupled to a linear quadrupole ion trap mass spectrometer, with complementary UV experiments performed with a benchtop OPO laser coupled to a cryogenically cooled 3D Paul ion trap.
2 Experimental methods
2.1 SOLEIL
The photo-processing of BNH+ under VUV irradiation was investigated using action spectroscopy with an LTQ linear ion trap (Milosavljević et al. 2012)coupled to the DESIRS undulator-based VUV beamline (Nahon et al. 2012) at the SOLEIL synchrotron facility in Saint-Aubin (France). Ions were generated by introducing a solution of 10 μM benzonitrile ( >99%, Sigma-Aldrich) diluted in acetonitrile into an atmospheric pressure photoionization source fitted with a krypton lamp. The cations were then guided through ion optics into the LTQ ion trap where a constant He pressure of ≈10−3 mbar was held, ensuring a good trapping efficiency and the thermalization of the ions at room temperature. BNH+ (m/z 104) was isolated through specific mass selection, preventing contamination from 13C isotopomers and possible fragments. Then, BNH+ was interrogated with the synchrotron radiation in the UV range from 4.5 to 7.5 eV in steps of 0.1 eV and in the VUV range from 7 to 13.6 eV in steps of 0.2 eV. Higher harmonics of the undulator synchrotron radiation were filtered out by a gas filter filled with Kr gas and with an additional Suprasil window when scanning the low energy region (4.5 to 7.5 eV). The monochromator exit slit width was set at 200 μm, leading to a photon resolution of 10 meV (at 10 eV) and a photon flux in the 1012 to 1013 photons s−1 range. The irradiation time was set at 500 ms to target a photo fragmentation yield less than 10% and avoid multiphoton sequential absorption processes. Tens of mass spectra were recorded at each photon energy and averaged to yield one mass spectrum per photon energy. The mass spectrometric data were treated using the MassJ package developed by Giuliani (2021) written in the Julia language (Bezanson et al. 2012). A statistical standard error can be retrieved from the averaging process as described in Giordano (2016) together with an error propagation treatment leading to the error bars given in the Fig. 2. The action spectra were normalized to the incident photon flux as measured by a calibrated VUV photodiode (IRD AXUV100), with a similar procedure as described by Wenzel et al. (2020).
2.2 PIIM
Photoexcitation of BNH+ from 3.5 to 5.5 eV was carried out in a cryogenically cooled Paul ion trap coupled with a tuneable ns UV-visible OPO laser (Alata et al. 2012; Esteves-López et al. 2015). Benzonitrile ( >99%, Sigma-Aldrich) was diluted in a solution of ~1:1 water:methanol to obtain a concentration of ≈10 μM. A few drops of acetic acid were added to promote protonation. Ions were generated using an electrospray ionization source and BNH+ was guided to a cryogenic Paul trap where it was trapped for several tens of ms. In that time, BNH+ was thermalised to a temperature of around 50 K through collisions with cold helium buffer gas. The photodissociation of the trapped ions was accomplished with a tuneable OPO laser (EKSPLA), which has a 10 Hz repetition rate, 10 ns pulse width, and a spectral width of ~10 cm−1 in the UV spectral region. The laser was shaped to a 1 mm2 spot to fit the entrance hole of the trap and the laser power was set around 2 mJ pulse−1. The precursor and photofragment product ions were extracted into a 1.5 m time-of-flight mass spectrometer after each laser shot and detected using a microchannel plates detector. The UV-visible photofragmentation spectra were recorded by detecting the ion signals as a function of the excitation energy.
3 Results
3.1 Photodissociation action spectrum
The experimental UV and VUV photodissociation action spectrum of BNH+ over the 3.5–9.6 eV range is shown in Fig. 1. The spectrum is obtained by summing all detected photoproducts. The solid purple trace corresponds to the spectrum obtained at PIIM laboratory by summing the major photoproduct m/z 77 and a minorcontribution of m/z 51 resulting from multiphoton processes. The dotted purple trace obtained at SOLEIL results from the sum of m/z 77 and a minor contribution of m/z 53. The solid green line is the calculated absorption spectra of BNH+ using TD-DFT method (Adamo & Jacquemin 2013) with the inclusion of 50 states at the ωB97XD/cc-pVTZ (Chai & Head-Gordon 2008; Dunning 1989) level of theory. Together, the experimental photodissociation spectra show four main bands centered at 4.3, 4.7, 6.4, and 7.6 eV after which the photofragment yield drops to zero around 9.5 eV before rising again slowly from 10.2 to 13.6 eV without exhibiting any major structure (see Fig. 2). The calculated spectrum of BNH+ has been shifted by −0.4 eV, to align with the main features of the experimental spectra. Only the spectral region exhibiting band structures in the experimental spectrum has been computed. The satisfactory agreement between the calculated absorption spectrum and the experimental photodissoication spectrum indicates that photodissociation is most likely the primary relaxation route upon UV/VUV photoexcitation. The vertical excitation energy of the first electronically allowed transition S1 ← S0 is calculated at 4.7 eV (4.3 eV in Fig. 1 after the −0.4 eV shift), indicating that the photodissociation experimental spectrum captures the first observable electronic transitions.
The photoproduct yield spectrum of BNH+ measured with an OPO laser at the PIIM laboratory (upper panel) onsets at ~3.8 eV and two broad features are apparent, centered at ~4.3 and ~4.7 eV. This is in good agreement with the study of Freiser & Beauchamp (1977), who reported a peak at 4.9 eV and a small shoulder at 4.2 eV using an lamp or monochromator source with a lower spectral resolution of ca. 10 nm. With the higher spectral resolution of our laser (minimum wavelength step 0.02 nm), we can confirm that the signal comprises two distinct bands. These are attributed to two electronic excited states, S1 and S2 in accord with the theoretical spectrum (lower panel). The S2 state is also probed in the photoproduct spectrum obtained at SOLEIL (central panel). The lack of vibronic structure in these electronic bands of BNH+ is notable since neutral BN exhibits vibrational structure in its first electronically excited state, beginning at 4.5 eV (273.88 nm, Kobayashi et al. 1987). Benzonitrile exhibits excited-state behaviour comparable to that of the interstellar species benzene: it has been shown that benzene also has vibronic structure in its neutral low-lying electronic excited states (Callomom et al. 1966)but not in its protonated forms (Freiser & Beauchamp 1977; Rode et al. 2009; Esteves-López et al. 2015).
 |
Fig. 1 Total photoproduct yield spectra of BNH+ measured atPIIM (solid purple) and at SOLEIL (dotted purple), obtained by summing all photoproducts. Theoretical absorption spectra of BNH+ (solid green line calculated by TD-DFT. The theoretical spectra are obtained by convoluting the stick spectra with Gaussian functions of 0.25 eV full width at half maximum and have been shifted by −0.4 eV. The final electronic excited states of the dominant electronic transitions for each band are labeled. |
 |
Fig. 2 VUV photodissociation action spectra of BNH+ over the 4.5–13.6 eV range, obtained by monitoring C6H |
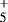
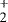
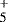
Dissociation thresholds for the dissociation channels of BNH+ obtained using zero-point corrected energies from CBS-QB3 calculations.
3.2 Fragmentation routes
The lowest calculated dissociation thresholds for BNH+ were obtained using the CBS-QB3 composite method (Wood et al. 2006) implemented in Gaussian 16 package (Frisch et al. 2016)and are presented in Table 1. The UV/VUV photodissociation action spectra of BNH+ depicted in Fig. 2, obtained at SOLEIL, show the presence of two dissociation channels. No formation of dications was observed experimentally in coherence with the adiabatic ionization energy of BNH+ calculated to be 13.96 eV (CBS-QB3 method). The main photodissociation channel corresponds to the neutral loss of 27 amu, as either HCN or HNC (black and blue traces in Fig. 2), and the minor dissociation channel correspond to the loss of a cyanoacetylene molecule (HC3N) plotted in red on a 5x zoomed scale. The m/z 77 channel (blue in Fig. 2) corresponds to the loss of HCN/HNC and the formation of C6 H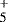 , which we assign as the phenylium cation. The phenylium cation is highly reactive towards water allowing us to assign the m/z 95 channel as the product of water addition to phenylium photoproduct cations. Water is present in background quantities in the ion trap. The change in the relative ratio of m/z 95 to m/z 77 signal along this energy range indicates different isomer populations of the m/z 77 cation. As all experimental conditions were held constant throughout acquisition of the spectrum, the ratio of m/z 95 to m/z 77 ions should be largely dominated by the reaction kinetics of the m/z 77 product and background H2 O. If the isomer distribution of m/z 77 changes, like from the onset of formation of linear species at higher photon energies, then this relative product ratio will change assuming that those linear species have different reactivity to the cyclic phenylium. The relative ratio of m/z95 to m/z 77 ions starts diminishing around 7 eV and we attribute this to the formation of a ring-opened species with a reduced reactivity towards background water.
, which we assign as the phenylium cation. The phenylium cation is highly reactive towards water allowing us to assign the m/z 95 channel as the product of water addition to phenylium photoproduct cations. Water is present in background quantities in the ion trap. The change in the relative ratio of m/z 95 to m/z 77 signal along this energy range indicates different isomer populations of the m/z 77 cation. As all experimental conditions were held constant throughout acquisition of the spectrum, the ratio of m/z 95 to m/z 77 ions should be largely dominated by the reaction kinetics of the m/z 77 product and background H2 O. If the isomer distribution of m/z 77 changes, like from the onset of formation of linear species at higher photon energies, then this relative product ratio will change assuming that those linear species have different reactivity to the cyclic phenylium. The relative ratio of m/z95 to m/z 77 ions starts diminishing around 7 eV and we attribute this to the formation of a ring-opened species with a reduced reactivity towards background water.
Figure 3 shows the predicted differences in electron density between our TD-DFT electronic excited states and the electronic ground state. In particular, S2 ←S0, S8 ←S0 and S12 ←S0 were chosen as they represent the dominant electronic transitions responsible for the three main peaks in the experimental photodissociation spectrum shown in Fig. 1. Electronic excitation to S2 and S8, where the C6 H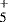 + H2O reaction is highly competitive, appear to be typical ππ* transitions. However, the transition to S12 completely disrupts the electronic π system and this could predispose the excited molecule to form a ring-open product.
+ H2O reaction is highly competitive, appear to be typical ππ* transitions. However, the transition to S12 completely disrupts the electronic π system and this could predispose the excited molecule to form a ring-open product.
The change in the reactivity of the m/z 77 product is visualised in Fig. 4, which shows the branching fractions into C6 H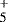 (blue triangles) and its water addition product (black circles), as well as the minor C4 H
(blue triangles) and its water addition product (black circles), as well as the minor C4 H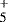 photoproduct channel (discussed later). The decrease in reactivity peaks around 8.5 eV and in the predicted region of the S12 ←S0 transition.
photoproduct channel (discussed later). The decrease in reactivity peaks around 8.5 eV and in the predicted region of the S12 ←S0 transition.
The minor dissociation channel ( ≈25 times weaker at 5 eV than the HCN/HNC loss channel) is assigned to the formation of C4 H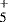 and neutral cyanoacetylene (HC3N). Although the branching ratio of m/z 53 is close to 0 from 4.5 to 10 eV, above this energy, it gradually increases to reach nearly 0.1 at 13.6 eV. The only isomer of C4 H
and neutral cyanoacetylene (HC3N). Although the branching ratio of m/z 53 is close to 0 from 4.5 to 10 eV, above this energy, it gradually increases to reach nearly 0.1 at 13.6 eV. The only isomer of C4 H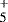 with a calculated product energy below the observed first band (4.7 eV) is the methyl-cyclopropyl cation at 4.40 eV, which corresponds to the global minimum on the C4 H
with a calculated product energy below the observed first band (4.7 eV) is the methyl-cyclopropyl cation at 4.40 eV, which corresponds to the global minimum on the C4 H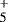 potential energy surface (Cunje et al. 1996; Muller et al. 2020).
potential energy surface (Cunje et al. 1996; Muller et al. 2020).
Wincel et al. (1990) and Chatterjee & Dopfer (2018) used mass spectrometry and infrared laser spectroscopy, respectively, to show that the preferred protonation site of benzonitrile is on the CN group. Furthermore, Wincel et al. (1990) demonstrated by combining mass spectrometry with isotope labeling that the H+ transfer from the CN group to the phenyl ring (preferentially to the “ortho” position) can occur at low internal energy and can even be followed by H-atom scrambling on the ring prior to ejection of a HCN molecule. Even if HCN loss is slightly thermodynamically favoured, compared to the HNC loss (Table 1), they have demonstrated that at higher internal energy the direct bond cleavage into C6H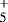 + HNC is preferred. In any case, the energy of the first electronic transition of BNH+ (4.3 eV) is above the product energy for both for HCN and HNC channels at 3.43 and 4.04 eV, respectively, so there is no information regarding the HCN/HNC neutral fragment.
+ HNC is preferred. In any case, the energy of the first electronic transition of BNH+ (4.3 eV) is above the product energy for both for HCN and HNC channels at 3.43 and 4.04 eV, respectively, so there is no information regarding the HCN/HNC neutral fragment.
 |
Fig. 3 Front (top) and side (bottom) view of the electron density difference maps for the electronic transitions predicted to be dominant for each of the three main features in the BNH+ photodissociation action spectrum. Blue indicates a region of increased electron density and red indicates areas of reduced electron density. We note that energies are given after the shift of −0.4 eV to best reproduce the experimental spectrum. |
 |
Fig. 4 UV/VUV dissociative branching ratios of BNH+. |
4 Astrophysical implications and conclusions
Although many routes for interstellar benzene formation have been proposed through radicals and molecular chemistry, the prevalent paths appear to be those involving ion–molecule reactions (Agúndez et al. 2021) and, notably, the one proposed by McEwan et al. (1999). The interstellar route to benzene formation proposed by McEwan et al. (1999) was based on a two-step mechanism from the phenylium cation. Firstly, a radiative association of C6 H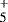 with H2 to form C6 H
with H2 to form C6 H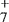 ,
,
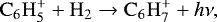 (1)
(1)
followed by dissociative recombination with an electron to lead to benzene,
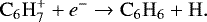 (2)
(2)
It is thus crucial to understand the formation routes of C6 H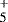 . Two main bottom-up pathways were proposed in the same study by McEwan et al. (1999):
. Two main bottom-up pathways were proposed in the same study by McEwan et al. (1999):
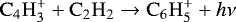 (3)
(3)
and
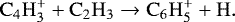 (4)
(4)
An additional path to C6 H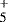 was also proposed by Herbst & Leung (1989),
was also proposed by Herbst & Leung (1989),
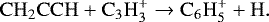 (5)
(5)
Based on ab initio molecular dynamics simulations, Peverati et al. (2016) showed that reaction (3) is a viable ion-molecule path to the formation of C6 H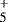 , which is itself a good nucleation center for the growth of larger aromatics in the ISM. Using reactions (1)–(3), Woods et al. (2002) calculated the column density of benzene to be within a factor of 2 of that observed in the protoplanetary nebula CRL 618 (Cernicharo et al. 2001). Interestingly, the model developed by Woods et al. (2002) also gave rise to a high abundance of benzonitrile, indicating that, as suggested by Burkhardt et al. (2021b), benzonitrile might be present in a large variety of interstellar and stellar regions.
, which is itself a good nucleation center for the growth of larger aromatics in the ISM. Using reactions (1)–(3), Woods et al. (2002) calculated the column density of benzene to be within a factor of 2 of that observed in the protoplanetary nebula CRL 618 (Cernicharo et al. 2001). Interestingly, the model developed by Woods et al. (2002) also gave rise to a high abundance of benzonitrile, indicating that, as suggested by Burkhardt et al. (2021b), benzonitrile might be present in a large variety of interstellar and stellar regions.
C6 H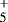 has been identified as a key molecule in PAH formation models, with the reactivity of phenylium being shown experimentally to produce more complex rings (Soliman et al. 2012). To date, no specific source of C6 H
has been identified as a key molecule in PAH formation models, with the reactivity of phenylium being shown experimentally to produce more complex rings (Soliman et al. 2012). To date, no specific source of C6 H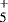 has been experimentally demonstrated despite its prominence as a bottleneck species in PAH formation (Jones et al. 2011), although it has been found to form at low abundances in plasma of simple hydrocarbons, including C2 H2 (Contreras & Salama 2013), and is postulated to form by sequential ion reaction of acetylene (Anicich et al. 2003).
has been experimentally demonstrated despite its prominence as a bottleneck species in PAH formation (Jones et al. 2011), although it has been found to form at low abundances in plasma of simple hydrocarbons, including C2 H2 (Contreras & Salama 2013), and is postulated to form by sequential ion reaction of acetylene (Anicich et al. 2003).
The photofragmentation identified here could represent a new “top-down” route to C6 H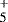 formation, unexplored in the extensive survey of “bottom-up” formation by Peverati et al. (2016). The authors of that study conclude that decay of C4 H
formation, unexplored in the extensive survey of “bottom-up” formation by Peverati et al. (2016). The authors of that study conclude that decay of C4 H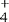 (formed via an ion-molecule reaction of acetylene) was the most likely route to form stable C4 H
(formed via an ion-molecule reaction of acetylene) was the most likely route to form stable C4 H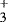 , whose subsequent reaction with C2 H2 yields stable C6 H
, whose subsequent reaction with C2 H2 yields stable C6 H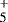 , a potential nucleus for PAH formation. Here, we show that UV/VUV photons can yield the product of this reaction pathway via the fragmentation of more complex aromatic species. The present observation that the phenylium cation is the major dissociation product of protonated benzonitrile under UV/VUV irradiation highlights the need for including formation paths of medium size species not only from bottom-up mechanisms but also from a top-down approach. Furthermore, the phenylium cation is highly reactive due to its extreme electrophilicity arising from its vacant nonbonding σ orbital, making it an appealing building block for larger aromatics in space (Ascenzi et al. 2007). The newly published detection of interstellar benzyne (o-C6H4) in TMC-1 further strengthens the argument for the characterization of reaction pathways yielding the phenylium cation, as C6 H
, a potential nucleus for PAH formation. Here, we show that UV/VUV photons can yield the product of this reaction pathway via the fragmentation of more complex aromatic species. The present observation that the phenylium cation is the major dissociation product of protonated benzonitrile under UV/VUV irradiation highlights the need for including formation paths of medium size species not only from bottom-up mechanisms but also from a top-down approach. Furthermore, the phenylium cation is highly reactive due to its extreme electrophilicity arising from its vacant nonbonding σ orbital, making it an appealing building block for larger aromatics in space (Ascenzi et al. 2007). The newly published detection of interstellar benzyne (o-C6H4) in TMC-1 further strengthens the argument for the characterization of reaction pathways yielding the phenylium cation, as C6 H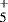 is predicted to undergo dissociative recombination with electrons to form benzyne (Cernicharo et al. 2021a).
is predicted to undergo dissociative recombination with electrons to form benzyne (Cernicharo et al. 2021a).
If we consider the specific case of Titan, solar irradiance is ~1% that on Earth, but the UV wing of the solar energy distribution incident upon the haze layer in the mesosphere drives a rich ionic and radical chemistry (Koskinen et al. 2011). Interestingly, the methyl-cyclopropyl cation resulting from the minor dissociation channel of BNH+ has been postulated to be responsible for the m/z 53 signal obtained by the mass spectrometer aboard the Cassini space probe while exploring Titan’s upper atmosphere (Peverati et al. 2016) and the mass corresponding to C6 H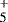 was measured during passes through the moon’s thermosphere (Vuitton et al. 2007). The neutral benzonitrile and BNH+ are currently integrated into the latest photochemical models of Titan’s atmosphere, as part of an “aromatic” fraction, where neutral benzonitrile is formed by the reaction of C6 H6 and the CN radical, with BNH+ formed by subsequent reaction with C2 H
was measured during passes through the moon’s thermosphere (Vuitton et al. 2007). The neutral benzonitrile and BNH+ are currently integrated into the latest photochemical models of Titan’s atmosphere, as part of an “aromatic” fraction, where neutral benzonitrile is formed by the reaction of C6 H6 and the CN radical, with BNH+ formed by subsequent reaction with C2 H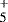 or HCNH+. Benzonitrile may also react with various species including C, CH, N2D or CN to form part of the “aromatic fraction” or photodissociate to form C6 H5, C6 H4 or C6 H4CN, while benzonitrile reforms from BNH+ via a reaction with, for instance, NH3, CH2NH, C3 H2, or with an electron (Loison et al. 2019). The chemical networks do not yet include the photoreactivity of BNH+ (Pearce et al. 2020), but in this work we have shown that its photofragmentation could represent a new, admittedly minor, route to C6 H
or HCNH+. Benzonitrile may also react with various species including C, CH, N2D or CN to form part of the “aromatic fraction” or photodissociate to form C6 H5, C6 H4 or C6 H4CN, while benzonitrile reforms from BNH+ via a reaction with, for instance, NH3, CH2NH, C3 H2, or with an electron (Loison et al. 2019). The chemical networks do not yet include the photoreactivity of BNH+ (Pearce et al. 2020), but in this work we have shown that its photofragmentation could represent a new, admittedly minor, route to C6 H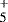 (and HCN/HNC). As such, it seems that the model of the photochemical production of aromatics in the atmosphere of Titan (Loison et al. 2019) could benefit from the addition of the photodissociation paths of charged benzonitrile. Furthermore, in the case of BNH+’s photodissociation, the neutral fragment ejected, HC3N is the smallest cyanopolyyne and also one of the key species in the chemical evolution of Titan’s atmosphere (Vuitton et al. 2007; Kaiser & Mebel 2012).
(and HCN/HNC). As such, it seems that the model of the photochemical production of aromatics in the atmosphere of Titan (Loison et al. 2019) could benefit from the addition of the photodissociation paths of charged benzonitrile. Furthermore, in the case of BNH+’s photodissociation, the neutral fragment ejected, HC3N is the smallest cyanopolyyne and also one of the key species in the chemical evolution of Titan’s atmosphere (Vuitton et al. 2007; Kaiser & Mebel 2012).
In conclusion, BNH+ participation in PAH growth is possible, albeit indirectly via the photofragmentation product C6 H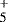 , which is already implicated in bottom-up PAH formation models (Burkhardt et al. 2021b). Since it is initiated by photofragmentation, the BNH+ contributionto PAH formation would need to be included in models – such as those applied to Titan’s atmosphere – as a two-step process, inevitably increasing the timescale of PAH growth through this mechanism. Most of the current astrochemical models do not account for the formation of C6 H
, which is already implicated in bottom-up PAH formation models (Burkhardt et al. 2021b). Since it is initiated by photofragmentation, the BNH+ contributionto PAH formation would need to be included in models – such as those applied to Titan’s atmosphere – as a two-step process, inevitably increasing the timescale of PAH growth through this mechanism. Most of the current astrochemical models do not account for the formation of C6 H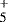 from the photodissociation of protonated benzonitrile, whereas C6 H
from the photodissociation of protonated benzonitrile, whereas C6 H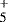 could play an important role in the chemical cycle as a benzonitrile precursor. This absence might partially rationalize the observed abundance of benzonitrile compared to predictions at a steady state. This work emphasizes the importance of obtaining absolute photodissociation cross-sections and branching ratios in the UV and VUV spectral range, which are to be incorporated into refined astrochemical models.
could play an important role in the chemical cycle as a benzonitrile precursor. This absence might partially rationalize the observed abundance of benzonitrile compared to predictions at a steady state. This work emphasizes the importance of obtaining absolute photodissociation cross-sections and branching ratios in the UV and VUV spectral range, which are to be incorporated into refined astrochemical models.
Acknowledgements
We are grateful to the staff from SOLEIL for the smooth running of the facility and providing beamtime under project 20191313. This work was partially supported by the Agence Nationale de la Recherche Scientifique, France, Project Numbers ANR-08-BLAN-0065, ANR2010BLANC040501-ESPEM and ANR17CE05000502-Wsplit, with additional support from the French Programme National “Physique et Chimie du Milieu Interstellaire” (PCMI) of the CNRS/INSU with the INC/INP, co-funded by the CEA and the CNES.
References
- Adamo, C., & Jacquemin, D. 2013, Chem. Soc. Rev., 42, 845 [CrossRef] [Google Scholar]
- Agúndez, M., Cabezas, C., Tercero, B., et al. 2021, A&A, 647, L10 [EDP Sciences] [Google Scholar]
- Alata, I., Omidyan, R., Broquier, M., Dedonder, C., & Jouvet, C. 2012, Chem. Phys., 399, 224 [NASA ADS] [CrossRef] [Google Scholar]
- Allamandola, L., Tielens, A., & Barker, J. 1989, ApJS, 71, 733 [NASA ADS] [CrossRef] [Google Scholar]
- Anicich, V. G., Wilson, P., & McEwan, M. J. 2003, J. Am. Soc. Mass Spectr., 14, 900 [Google Scholar]
- Ascenzi, D., Cont, N., Guella, G., Franceschi, P., & Tosi, P. 2007, J. Phys. Chem. A, 111, 12513 [CrossRef] [Google Scholar]
- Bezanson, J., Karpinski, S., Shah, V. B., & Edelman, A. 2012, ArXiv eprints [arXiv:1209.5145] [Google Scholar]
- Burkhardt, A. M., Lee, K. L. K., Changala, P. B., et al. 2021a, ApJ, 913, L18 [Google Scholar]
- Burkhardt, A. M., Loomis, R. A., Shingledecker, C. N., et al. 2021b, Nat. Astron., 5, 181 [Google Scholar]
- Callomom, J., Dunn, T., & Mills, I. M. 1966, Philos. Trans. R. Soc. A, 259, 499 [NASA ADS] [Google Scholar]
- Campbell, E. K., Holz, M., Gerlich, D., & Maier, J. P. 2015, Nature, 523, 322 [NASA ADS] [CrossRef] [Google Scholar]
- Cernicharo, J., Heras, A. M., Tielens, A., et al. 2001, ApJ, 546, L123 [NASA ADS] [CrossRef] [Google Scholar]
- Cernicharo, J., Agúndez, M., Kaiser, R. I., et al. 2021a, A&A, 652, L9 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cernicharo, J., Agúndez, M., Cabezas, C., et al. 2021b, A&A, 649, L15 [EDP Sciences] [Google Scholar]
- Chai, J.-D., & Head-Gordon, M. 2008, Phys. Chem. Chem. Phys., 10, 6615 [NASA ADS] [CrossRef] [Google Scholar]
- Chatterjee, K., & Dopfer, O. 2018, ApJ, 865, 114 [NASA ADS] [CrossRef] [Google Scholar]
- Cherchneff, I., Barker, J. R., & Tielens, A. G. 1992, ApJ, 401, 269 [NASA ADS] [CrossRef] [Google Scholar]
- Contreras, C. S., & Salama, F. 2013, ApJS, 208, 6 [NASA ADS] [CrossRef] [Google Scholar]
- Coustenis, A., Salama, A., Schulz, B., et al. 2003, Icarus, 161, 383 [CrossRef] [Google Scholar]
- Cunje, A., Rodriquez, C., Lien, M., & Hopkinson, A. 1996, J. Org. Chem., 61, 5212 [Google Scholar]
- Delitsky, M. L., & McKay, C. P. 2010, Icarus, 207, 477 [CrossRef] [Google Scholar]
- Dunning, Jr, T. H. 1989, J. Chem. Phys., 90, 1007 [NASA ADS] [CrossRef] [Google Scholar]
- Esteves-López, N., Dedonder-Lardeux, C., & Jouvet, C. 2015, J. Chem. Phys., 143, 074303 [CrossRef] [Google Scholar]
- Etim, E. E., Gorai, P., Das, A., & Arunan, E. 2017, Adv. Space Res., 60, 709 [NASA ADS] [CrossRef] [Google Scholar]
- Freiser, B., & Beauchamp, J. 1977, J. Am. Chem. Soc., 99, 3214 [CrossRef] [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. 2016, Gaussian~16 Revision C.01, gaussian Inc. Wallingford CT [Google Scholar]
- Geballe, T., & Oka, T. 1996, Nature, 384, 334 [NASA ADS] [CrossRef] [Google Scholar]
- Geballe, T., McCall, B., Hinkle, K., & Oka, T. 1999, ApJ, 510, 251 [CrossRef] [Google Scholar]
- Giordano, M. 2016, ArXiv e-prints [arXiv:1610.08716] [Google Scholar]
- Giuliani, A. 2021, MassJ : Julia package for mass spectrometry data treatment and analysis. https://doi.org/10.15454/ku1p28 [Google Scholar]
- Herbst, E., & Leung, C. M. 1989, ApJS, 69, 271 [Google Scholar]
- Jones, B. M., Zhang, F., Kaiser, R. I., et al. 2011, Proc. Natl. Acad. Sci., 108, 452 [Google Scholar]
- Kaiser, R. I., & Mebel, A. M. 2012, Chem. Soc. Rev., 41, 5490 [Google Scholar]
- Khare, B., Sagan, C., Zumberge, J. E., Sklarew, D. S., & Nagy, B. 1981, Icarus, 48, 290 [NASA ADS] [CrossRef] [Google Scholar]
- Kobayashi, T., Honma, K., Kajimoto, O., & Tsuchiya, S. 1987, J. Chem. Phys., 86, 1111 [NASA ADS] [CrossRef] [Google Scholar]
- Koskinen, T., Yelle, R., Snowden, D., et al. 2011, Icarus, 216, 507 [CrossRef] [Google Scholar]
- Larsson, M., Geppert, W., & Nyman, G. 2012, Rep. Prog. Phys., 75, 066901 [NASA ADS] [CrossRef] [Google Scholar]
- Lee, K. L. K., McGuire, B. A., & McCarthy, M. C. 2019, Phys. Chem. Chem. Phys., 21, 2946 [CrossRef] [Google Scholar]
- Lee, K. L. K., Changala, P. B., Loomis, R. A., et al. 2021, ApJ, 910, L2 [NASA ADS] [CrossRef] [Google Scholar]
- Lipkus, A. H., Yuan, Q., Lucas, K. A., et al. 2008, J. Org. Chem., 73, 4443 [Google Scholar]
- Loison, J., Dobrijevic, M., & Hickson, K. 2019, Icarus, 329, 55 [CrossRef] [Google Scholar]
- McCall, B., Geballe, T., Hinkle, K., & Oka, T. 1998, Science, 279, 1910 [CrossRef] [Google Scholar]
- McCall, B., Geballe, T., Hinkle, K., & Oka, T. 1999, ApJ, 522, 338 [NASA ADS] [CrossRef] [Google Scholar]
- McCarthy, M. C., Lee, K. L. K., Loomis, R. A., et al. 2021, Nat. Astron., 5, 176 [Google Scholar]
- McEwan, M. J., Scott, G. B., Adams, N. G., et al. 1999, ApJ, 513, 287 [CrossRef] [Google Scholar]
- McGuire, B. A., Burkhardt, A. M., Kalenskii, S., et al. 2018, Science, 359, 202 [Google Scholar]
- McGuire, B. A., Loomis, R. A., Burkhardt, A. M., et al. 2021, Science, 371, 1265 [Google Scholar]
- Milosavljević, A. R., Nicolas, C., Gil, J.-F., et al. 2012, J. Synchrotron Rad., 19, 174 [CrossRef] [Google Scholar]
- Muller, G., Jacovella, U., Catani, K. J., da Silva, G., & Bieske, E. J. 2020, J. Phys. Chem. A, 124, 2366 [NASA ADS] [CrossRef] [Google Scholar]
- Nahon, L., de Oliveira, N., Garcia, G. A., et al. 2012, J. Synchrotron Rad., 19, 508 [Google Scholar]
- Nixon, C. A., Thelen, A. E., Cordiner, M. A., et al. 2020, AJ, 160, 205 [NASA ADS] [CrossRef] [Google Scholar]
- Pearce, B. K., Molaverdikhani, K., Pudritz, R. E., Henning, T., & Hébrard, E. 2020, ApJ, 901, 110 [NASA ADS] [CrossRef] [Google Scholar]
- Peverati, R., Bera, P. P., Lee, T. J., & Head-Gordon, M. 2016, ApJ, 830, 128 [CrossRef] [Google Scholar]
- Rode, M. F., Sobolewski, A. L., Dedonder, C., Jouvet, C., & Dopfer, O. 2009, J. Phys. Chem. A, 113, 5865 [NASA ADS] [CrossRef] [Google Scholar]
- Schlemmer, S., Cook, D., Harrison, J., et al. 1994, Science, 265, 1686 [NASA ADS] [CrossRef] [Google Scholar]
- Soliman, A.-R., Hamid, A. M., Momoh, P. O., et al. 2012, J. Phys. Chem. A, 116, 8925 [NASA ADS] [CrossRef] [Google Scholar]
- Vuitton, V., Yelle, R., & McEwan, M. 2007, Icarus, 191, 722 [NASA ADS] [CrossRef] [Google Scholar]
- Wenzel, G., Joblin, C., Giuliani, A., et al. 2020, A&A, 641, A98 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Wincel, H., Fokkens, R., & Nibbering, N. 1990, J. Am. Soc. Mass Spectr., 1, 225 [NASA ADS] [CrossRef] [Google Scholar]
- Wood, G. P., Radom, L., Petersson, G. A., et al. 2006, J. Chem. Phys., 125, 094106 [NASA ADS] [CrossRef] [Google Scholar]
- Woods, P. M., Millar, T., Zijlstra, A., & Herbst, E. 2002, ApJ, 574, L167 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
Dissociation thresholds for the dissociation channels of BNH+ obtained using zero-point corrected energies from CBS-QB3 calculations.
All Figures
|
Fig. 1 Total photoproduct yield spectra of BNH+ measured atPIIM (solid purple) and at SOLEIL (dotted purple), obtained by summing all photoproducts. Theoretical absorption spectra of BNH+ (solid green line calculated by TD-DFT. The theoretical spectra are obtained by convoluting the stick spectra with Gaussian functions of 0.25 eV full width at half maximum and have been shifted by −0.4 eV. The final electronic excited states of the dominant electronic transitions for each band are labeled. |
| In the text | |
|
Fig. 2 VUV photodissociation action spectra of BNH+ over the 4.5–13.6 eV range, obtained by monitoring C6H |
| In the text | |
|
Fig. 3 Front (top) and side (bottom) view of the electron density difference maps for the electronic transitions predicted to be dominant for each of the three main features in the BNH+ photodissociation action spectrum. Blue indicates a region of increased electron density and red indicates areas of reduced electron density. We note that energies are given after the shift of −0.4 eV to best reproduce the experimental spectrum. |
| In the text | |
|
Fig. 4 UV/VUV dissociative branching ratios of BNH+. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.