| Issue |
A&A
Volume 633, January 2020
|
|
|---|---|---|
| Article Number | A103 | |
| Number of page(s) | 6 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/201936873 | |
| Published online | 17 January 2020 | |
Fragmentation and isomerization of polycyclic aromatic hydrocarbons in the interstellar medium: Coronene as a case study
1
School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Theoretical Chemistry and Biology, Royal Institute of Technology, 10691 Stockholm, Sweden
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
, This email address is being protected from spambots. You need JavaScript enabled to view it.
2
Hefei National Laboratory for Physical Science at the Microscale, Department of Chemical Physics, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, 230026 Anhui, PR China
3
Department of Physics and Astronomy, University of Missouri, Columbia, MO 65211, USA
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
8
October
2019
Accepted:
15
December
2019
Abstract
Aims. Due to the limitations of current computational technology, the fragmentation and isomerization products of vibrationally-excited polycyclic aromatic hydrocarbon (PAH) molecules and their derivatives have been poorly studied. In this work, we investigate the intermediate products of PAHs and their derivatives as well as the gas-phase reactions relevant to the interstellar medium, with coronene as a case study.
Methods. Based on the semi-empirical method of PM3 as implemented in the CP2K program, molecular dynamics simulations were performed to model the major processes (e.g., vibrations, fragmentations, and isomerizations) of coronene and its derivatives (e.g., methylated coronene, hydrogenated coronene, dehydrogenated coronene, nitrogen-substituted coronene, and oxygen-substituted coronene) at temperatures of 3000 K and 4000 K.
Results. We find that the anharmonic effects are crucial for the simulation of vibrational excitation. For the molecules studied here, H2, CO, HCN, and CH2 are the major fragments. Following the dissociation of these small units, most of the molecules could maintain their ring structures, but a few molecules would completely break into carbon chains. The transformation from a hexagon to a pentagon or a heptagon may occur and the heteroatomic substitutions (e.g., N- or O-substitutions) would facilitate the transformation.
Key words: astrochemistry / molecular data / molecular processes / ISM: molecules / methods: laboratory: molecular / photon-dominated region
© ESO 2020
1. Introduction
Polycyclic aromatic hydrocarbon (PAH) molecules are commonly thought to be responsible for the distinctive set of aromatic infrared (IR) emission features at 3.3, 6.2, 7.7, 8.6, and 11.3 μm, which are seen in a wide variety of astrophysical environments (Leger & Puget 1984; Allamandola et al. 1985). In the interstellar medium (ISM), PAHs are vibrationally excited by ultraviolet (UV) and visible stellar photons (Li & Draine 2002). Following the photoexcitation, electronic relaxation predominantly takes place by nonradiative processes in which the intramolecular vibrational energies are redistributed randomly over all accessible vibrational degrees of freedom of the electronic ground state of the molecules (Allamandola et al. 1989). The vibrational de-excitation occurs from these vibrationally hot molecules, which lead to the aromatic IR emission bands (Tielens 2008). Besides the IR emission, fragmentation is a competing process for a vibrationally excited molecule. However, the timescales are very different for these two processes. The fragmentation, which resulted from photo-driven processes, takes place in a timescale of picoseconds (ps), and the loss of H, H2, or C2nHx usually dominates the experimental mass spectra of PAH fragmentation (West et al. 2014; Chen et al. 2018; Zhen et al. 2018). Since the redistribution of the absorbed energy and the subsequent fragmentations are random, such a process is usually called statistical fragmentation (Chen et al. 2014, 2015).
In addition to photo-driven processes, PAHs are also subjected to energetic ions from stellar winds or supernova explosions (Micelotta et al. 2010). As a result of this astrophysical interest, the ion-induced fragmentation and molecular growth processes have recently been extensively investigated (Zettergren et al. 2013; Seitz et al. 2013; Stockett et al. 2014a,b; Chen et al. 2014, 2015; Delaunay et al. 2015). At low energies, with a center-of-mass energy less than 1 keV, energetic ions lead to the loss of H and C atoms. The chemical routes and fragmentation products are expected to appreciably differ from that of photo-driven processes (i.e., the statistical fragmentation). This process is called nonstatistical fragmentation (Stockett et al. 2014b; Chen et al. 2014, 2015), in which, single C-losses are commonly observed (Stockett et al. 2014a,b, 2015). Nonstatistical fragmentation occurs approximately a thousand times faster than the statistical fragmentation, that is, at a timescale of femtoseconds (fs).
At high center-of-mass energies (≳10 keV) of ion-induced dissociation, the statistical fragmentation is dominant, that is, H-, H2-, and C2nHx-losses are most commonly detected on the experimental mass spectra (Zettergren et al. 2013; Seitz et al. 2013; Delaunay et al. 2015; Chen et al. 2015). This process is crucial for a PAH molecule to reach high internal energy or temperature, which cannot be reached through the absorption of single photons below the Lyman limit. The main drawback with the experiments is that only the mass-to-charge ratios can be measured and no detailed information about the products, the intermediate fragments, nor the reaction processes can be recorded. To compensate for this drawback, static quantum chemical calculations, such as, density functional theory (DFT), are performed to reveal the possible structures and reactions associated with a product. The reaction pathways for the formation of H and H2 from PAHs have been revealed through static DFT (Paris et al. 2014; Chen et al. 2015; Castellanos et al. 2018). However, the static methods rely heavily on the empirical knowledge about complex systems and reactions, for example, the mechanism for C2nHx-losses or complete fragmentation of carbon skeletons can hardly be explored by using the static methods. Instead, molecular dynamics simulations are widely performed to investigate the detailed fragmentation processes (Martín-Sómer et al. 2016; Krasnokutski et al. 2017; Trinquier et al. 2017; Simon et al. 2018; Rapacioli et al. 2018; Chen & Luo 2019).
In this work, we apply molecular dynamics simulations to investigate the vibrations, H-migrations, dissociations, and isomerizations of PAHs, with coronene (C24H12), a highly symmetric PAH species, as a case study1.
In astronomical environments, PAH molecules may include substituents, such as N in place of C (e.g., see Hudgins et al. 2005; Mattioda et al. 2008) and O in place of C (e.g., see Bauschlicher 1998). Observationally, the C–C stretch peaks at wavelengths as short as 6.2 μm, in contrast, both experimental and computational spectra of pure PAHs reveal that it often occurs at ≳6.3 μm. The subtle variations in the peak wavelength of the 6.2 μm C–C emission band are commonly attributed to polycyclic aromatic nitrogen heterocycles; PAHs with one or more nitrogen atoms that are substituted into their carbon skeleton (Hudgins et al. 2005; Mattioda et al. 2008). In regions with intense UV radiation, small PAHs with fewer than 25 carbon atoms are expected to be partially dehydrogenated (e.g., see Tielens et al. 1987; Malloci et al. 2008)2. In H-rich, UV-poor benign regions, PAHs may be superhydrogenated and their edges contain excess H atoms (e.g., see Bernstein et al. 1996; Thrower et al. 2012; Sandford & Bernstein 2013). They may also contain one or several methyl sidegroups as revealed by the detection in many PAH sources of a weak satellite emission feature at 3.4 μm, which always accompanies the 3.3 μm emission feature (see Yang et al. 2017 and references therein). Finally, experiments have shown that UV photolysis of low-temperature coronene-ice mixtures in dense molecular clouds leads to the addition to PAHs of various functional groups (Bernstein et al. 1999, 2002; Gibb et al. 2000), including methyl (–CH3), methoxy (–OCH3), cyano (–CN), isocyano (–NC), alcohol (–OH), and ketone (> C=O).
To explore the influence of the structural complexities of PAHs on their fragmentation and isomerization processes, we therefore also examine seven derivatives of coronene, including methylated coronene, hydrogenated coronene, dehydrogenated coronene, N-substituted coronene, O-substituted coronene, carbonylcoronene, and methoxycoronene (see Fig. 1). This paper is organized as follows. Section 2 shows the importance of the anharmonic effects; neither dissociation nor isomerization could be accurately studied if these effects are not taken into account. The various relevant fragmentation processes are elaborated on in Sect. 3. In Sect. 4 we show a new H roaming route. Section 5 explores the formation of a pentagon and a heptagon in PAH derivatives. Finally, we summarize the major results in Sect. 6.
 |
Fig. 1. Structures of coronene and its seven derivatives studied in this work. The names and chemical formulae are shown beneath each molecule. |
2. Anharmonicity
For harmonic oscillators, the vibrational energy levels on the parabolic potential energy surface are equally separated (see Fig. 2 for an illustration), which can be calculated from the following:
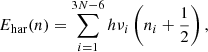 (1)
(1)
 |
Fig. 2. Harmonic and anharmonic potentials. Harmonic potentials can be described by a parabolic function, which is a symmetric well, that closes on both sides. A molecule does not break up with such a harmonic potential. Anharmonic potential is an asymmetric and open well. At certain vibrational states, a molecule dissociates. |
where h is the Planck constant; νi is the frequency of the ith vibrational mode, for a nonlinear molecule of N atoms, there are 3N − 6 vibrational modes; and ni ≡ (n1, n2,...) is the quantum number representing the state of each vibrational mode. The fundamental vibrational frequency of a molecule corresponds to the transition from the ground level (n = 0) to the first vibrational excitation level (n = 1). The zero point energy is Ehar(0) = (1/2)∑ihνi. The transitions from n = 0 to m > 1 are called overtones, and the transitions from n ≥ 1 to m > n are called hot transitions or hot bands.
According to Eq. (1), a molecule with harmonic bonds could reach infinite energy Ehar(n → ∞) without bond breaking. However, this is not the case in reality. Molecules do dissociate at highly excited vibrational states (Chen et al. 2015; Chen & Luo 2019). The potential energy surface is not a perfectly parabolic shape, instead, it is a non-symmetric open well (i.e., anharmonic potential, see Fig. 2). This allows a molecule to break up above certain vibrational states. On the anharmonic potential energy surface, the energy levels are unequally separated (see Fig. 2 for illustration), which can be estimated from the following formula (Burcl et al. 2003):
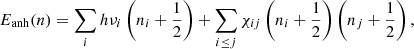 (2)
(2)
where χij, which is the anharmonic coupling that describes the interactions (mode couplings) or resonances among various vibrational modes, can be approximated as a two-dimensional matrix, and most of its elements are negative. This leads to the anharmonic energy levels Eanh(n) that are lower than the corresponding harmonic energy levels Ehar(n). Moreover, due to mode couplings, combination bands of two or more fundamental vibrations that are excited simultaneously show up, and the positions and intensities of the fundamental bands also change (Chen 2018). The band shifting and broadening have been seen in the vibrational spectra, which were calculated by molecular dynamics simulations (Chen 2019), suggesting that anharmonicity and mode couplings are intrinsically included in the molecular dynamics simulations.
3. Fragmentation
Figure 4 shows the experimental mass spectrum of coronene following collisions with He+ (Chen et al. 2015). Large amounts of H losses and fragmentations of the carbon skeleton are clearly seen. The mechanism for H losses has been studied in detail (Chen et al. 2015; Castellanos et al. 2018). However, due to the complex isomerization and dissociation features at highly excited vibrational states, the reaction pathway for the fragmentation of the carbon skeleton remains uncharted. Here, we investigate such reactions using molecular dynamics simulations.
It has been reported that the fragmentation only becomes important for temperatures exceeding ∼ 2200 K, regardless of the PAH size and the excitation agent (Chen et al. 2015). Therefore, we simulated vibrational excitations starting from 3000 K. We should note that, as shown in Fig. 3, for a coronene molecule at a vibrational temperature of T ≳ 3000 K, its vibrational energy has to exceed ∼ 20 eV. This indicates that the fragmentation process described here would be mostly applicable to UV-intense regions where PAHs are excited by multiple photons or hostile environments where PAHs are collisionally excited by energetic particles. As the energy can be randomly redistributed across all degrees of freedom, such simulations correspond to statistical fragmentations. The simulations were performed using the Quickstep module of the CP2K program package (VandeVondele et al. 2005). The semi-empirical method of PM3 (Stewart 1989) was utilized for electronic structure calculations. It has been shown that such methods are capable of producing reasonable vibrational spectra for large compact PAHs (Chen 2019) and high-temperature dissociation pathways for linear PAHs (Chen & Luo 2019). Canonical ensembles (NVT) were performed at multiple temperatures with the Nosé–Hoover chain thermostat (Nosé 1984a,b; Martyna et al. 1992). Starting from optimized geometries of the individual molecules, the systems were equilibrated for 10 ps. The production simulations were subsequently run for 50 ps, at a time step of 0.1 fs.
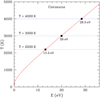 |
Fig. 3. Temperature T for coronene with vibrational energy E, calculated from its vibrational mode spectrum (see Draine & Li 2001). For coronene to attain a vibrational temperature of T ≳ 2200, 3000, and 4000 K, its vibrational energy E has to exceed ∼13.2, 20.0, and 28.5 eV, respectively. This can only be achieved for PAHs excited by multiple photons or energetic particles. |
 |
Fig. 4. Mass spectrum of coronene following collisions with 11.25 keV He+. The highest peaks correspond to the single and double ionizations of coronene without fragmentation on the experimental time scale of ∼10 μs. The left and right zoom-ins respectively show the intensity distributions for the losses of different numbers, n, of H atoms from the coronene dications and cations. The loss of an even number of H atoms is more favorable than the loss of an odd number of H atoms. Other peaks are due to the fragmentation of the carbon skeleton in one or several steps. See Chen et al. (2015) for details. |
Figure 5 shows the snapshots from molecular dynamics simulations for 1-hydrocoronene and O-substituted coronene at 3000 K. At this temperature, no fragment is observed for other molecules. As demonstrated in Fig. 5, due to the addition of an extra H atom to a peripheral carbon, the corresponding C–H bond converts from aromatic to aliphatic, which weakens the strength of the connection and eventually leads to the first break up of the C–H bond at 0.28 ps. The hydrogenated coronene is less stable in comparison to a pristine coronene, which breaks up the first C–H bond at 4000 K. This phenomenon is consistent with a recent finding that additional H atoms do not protect PAH molecules from fragmentation (Gatchell et al. 2015). At 0.54 ps a hydrogen atom migrates from the body of the newly formed carbon chain to the unsaturated carbon, and at 0.55 ps a new C–H bond is formed. At 1.26 ps, a hydrogen atom on the head of the carbon chain is dissociated, that is, sp3 C–C or sp2 C=C bonds tend to form sp C≡C bonds by removing H atoms (Chen & Luo 2019). At the end of the simulation (50 ps), a smaller PAH with only one broken ring attached to a –C2H sidegroup that formed, which agrees with the results reported recently for linear PAHs (Chen & Luo 2019).
 |
Fig. 5. Fragmentation of 1-hydrocoronene (top) and O-substituted coronene (bottom) at 3000 K. The orange text in the cells indicates the fragment(s) which are located far away from the maternal molecule. |
For the O-substituted coronene, one of the C–O bonds breaks up at first at 8.08 ps. Similar to 1-hydrocoronene, the hydrogen atom that is linked to the other side of the C–O bond migrates to the unsaturated carbon to form a new C–H bond. A CO molecule is formed at 13.82 ps and dissociates from the maternal molecule. No other fragment is observed and no ring is broken until the end of the simulation (50 ps). This process is consistent with the experimental and theoretical studies of the photodissociation of bisanthenequinone cations in which the CO-loss is found to be the lowest dissociation channel (Chen et al. 2018).
The other molecules dissociate at 4000 K. Again, as shown in Fig. 3, such a high vibrational temperature can only be achievable for PAHs that are vibrationally excited by multiple photons or energetic particles. Figure 6 shows the fragmentation processes of coronene, 1-methylcoronene, coronene with a H-loss, N-substituted coronene, coronene with a hydrogen atom substituted by an oxygen atom, and 1-methoxycoronene. For coronene, a H2-loss is observed, it is very similar to the reaction pathway as reported in the literature, that is, H2 is formed via H migration without breaking the ring (Paris et al. 2014; Chen et al. 2015; Castellanos et al. 2018). For 1-methylcoronene, the C–CH3 bond breaks at 0.25 ps. In the meantime, one of the hydrogen atoms on the methyl group moves to the carbon atom that is connected with –CH3. At 0.31 ps, a bare coronene molecule is formed and a CH2-loss is observed simultaneously. No more fragment is found until the end of the simulation. However, one of the peripheral hexagons tranforms to a pentagon attached with a –CH2 sidegroup (see the last cell in the second row of Fig. 6 for details).
 |
Fig. 6. Fragmentation of (from top to bottom) coronene, 1-methylcoronene, coronene with a H-loss, nitrogen-substituted coronene, 1-carbonylcoronene, and 1-methoxycoronene. The simulations were performed at 4000 K. The orange text(s) in the cells indicate the fragment(s) which are located far away from the maternal molecule. |
Both coronene and 1-methylcoronene maintain the pristine aromatic structures, that is, most of the rings do not break at 4000 K. However, the situation is different for the coronene with a H-loss. At 1.46 ps, three rings are broken into carbon chains, and a hydrogen atom is released. At 3.58 ps, all the rings are broken, and a C5H2 molecule and a longer carbon chain are dissociated from each other. Subsequently, a H-loss from the longer chain is observed at 5.22 ps. At 6.59 ps, a C3H2 molecule is separated from the longer chain. The molecule is completely fragmented into multiple carbon chains and H atoms.
For the N-substituted coronene, HCN is formed at 0.425 ps. No more fragments are found until the end of the simulation. However, the maternal molecule isomerizes to a smaller PAH attached with carbon chains. The fragmentation of the coronene with a hydrogen atom that is substituted by an oxygen atom is also violent. At 0.585 ps, five rings are broken, and a CO unit is released from the main body. At 0.815 ps, a hydrogen atom is dissociated. At 1.275 ps, a C5H2 molecule is separated. Similar to the case for the coronene with a H-loss, the molecule is completely dissociated to multiple carbon chains, CO, and H atoms. 0 One-methoxycoronene is the largest molecule in this study, which contains 40 atoms. At 6.66 ps, a H2 molecule is released from the methoxyl group. At 15.07 ps, another H2 molecule is dissociated and the carbon atom on the methoxyl group migrates from the oxygen atom to a neighboring carbon atom, that is, a –CO unit is formed. As previously mentioned, for both O-substituted coronene and 1-carbonylcoronene, the C–CO bond always breaks before C–H and C–C bonds. At 21.84 ps, a CO-loss is observed. Thereafter, no more fragments are found and no rings break until the end of the simulation.
4. Hydrogen roaming
The roaming of hydrogen atoms is crucial for the formation of aliphatic bonds and H2 molecules (Paris et al. 2014; Chen et al. 2015; Castellanos et al. 2018). Previous studies have reported hydrogen roaming on the periphery of PAHs. In this study, we find a new roaming route. Figure 7 shows the hydrogen roaming on the carbon skeleton of N-substituted coronene. The following two hydrogen migrations are demonstrated: at 4.53 ps, one hydrogen atom is located at the end of a carbon chain, the other one is bound to an inner carbon atom that belongs to a pentagon. At 4.545 ps, the carbon atom located at the end of the carbon chain dissociates from the carbon chain, and at 4.55 ps, it migrates to a nearby unsaturated carbon atom that belongs to a hexagon. At 4.62 ps, the hydrogen atom located at the inner carbon atom migrates to the carbon chain, which transfers a hydrogen atom to a nearby carbon atom at 4.55 ps. At 4.635 ps, all the peripheral carbon atoms become saturated with hydrogen atoms.
 |
Fig. 7. Hydrogen roaming on C22H10 fragment produced by ejection of HCN from nitrogen-substituted coronene. |
5. Formation of pentagons and heptagons
Figure 8 shows the formation of pentagons in the photodissociation of bisanthenequinone cations (Bq+), in which the Bq+ cation does not dehydrogenate. Instead, it fragments via the sequential loss of two neutral carbonyl groups (–CO), causing the formation of pentagons. In addition, the molecule transforms into a bowl-shaped one following the loss of the second –CO group. This process is proposed to be an important step for the formation of fullerene in the ISM (Chen et al. 2018). Similar reactions can also be found in Figs. 6 and 7.
 |
Fig. 8. Mass spectrum of bisanthenequinone cation (Bq+) following irradiation by laser. The three major peaks represent Bq+, [Bq-CO]+, and [Bq-2CO]+, respectively (from right to left). The optimized structures of the molecules are shown beside each peak. See Chen et al. (2018) for details. |
Figure 9 shows another example of pentagon formation in the N-substituted coronene. At 0.325 ps, a HCN molecule is dissociated from the maternal molecule, which opens two hexagons, and forms two short carbon chains. At 0.335 ps, one carbon chain closes up to form a pentagon. Then at 0.395 ps another pentagon is formed due to the close up of the other carbon chain. These processes are similar to the formation of pentagons in Bq+(Chen et al. 2018). Recent work has also shown that the loss of a HCN molecule in nitrogen-containing PAHs offers a facile pathway toward pentagon formation (de Haas et al. 2017).
 |
Fig. 9. Formation of pentagons in nitrogen-substituted coronene. |
Apart from pentagons, we find the formation of a heptagon in 1-methoxycoronene. Following the loss of two H2 molecules, an unsaturated carbon (C=C) and a –CO functional group are formed (see Fig. 10 and the last line in Fig. 6). At 19.5 ps, the bond linking the C=C and –CO units breaks. As shown above, the unsaturated carbon is very reactive, which always tends to form covalent bonds with a neighboring atom. At 19.53 ps, the unsaturated carbon atom bends toward the neighboring carbon atom, and at 19.54 ps, a heptagon is formed.
 |
Fig. 10. Formation of heptagons in 1-methoxycoronene. |
6. Concluding remarks
To date, more than 200 molecules have been identified in the ISM. To understand the origin and evolution of astronomically-relevant molecules, the time-of-flight (TOF) mass spectroscopy experiments in combination with the static quantum chemical calculations are commonly performed. However, this combination can hardly predict intermediate or temporary products of the studied molecules, especially when the molecules are vibrationally excited by photons, electrons, or stellar winds from nearby stars. In this work, by using molecular dynamics simulations, various fragmentation and isomerization features of coronene and its seven functionalized derivatives are investigated at high temperatures. It is found that most of these molecules do not dissociate at 3000 K, except hydrogenated coronene and oxygen-substituted coronene. These two molecules release hydrogen atoms and CO units at 3000 K. Due to the loss of hydrogen, one of the peripheral rings in hydrogenated coronene breaks, that is, a six-ring that is attached with a –C2H unit structure is formed. However, the structure of the oxygen-substituted coronene remains closed (i.e., no open rings) following the loss of CO. The other six molecules dissociate at 4000 K: (i) bare coronene releases a H2 molecule without breaking any rings; (ii) 1-methylcoronene releases a CH2 unit and forms a pentagon that is bonded with a –CH2 unit at the end of the simulation; (iii) dehydrogenated coronene (i.e., coronene with a H atom loss) completely breaks into H atoms and carbon chains; (iv) nitrogen-substituted coronene releases a HCN unit and breaks two rings; (v) coronene with a H atom substituted by an oxygen atom dissociates to H, CO, and carbon chains of various sizes; and (vi) 1-methoxycoronene releases two H2 molecules and one CO unit, however, no ring breaks until the end of the simulation. The ring structures with a carbon-chain that is attached and the formations of pentagons and heptagons are commonly observed in the simulations, especially in heteroatomic substituted molecules. The heteroatomic substitutions (e.g., nitrogen- and oxygen-substitutions) play a key role in the formation of pentagons or heptagons. We note that the vibrational temperatures of 3000 K and 4000 K that are examined in this work cannot be easily achieved by small PAHs through the absorption of single stellar photons (see Fig. 13 of Draine & Li 2001 and Fig. 4 of Draine & Li 2007). For coronene to attain such high vibrational temperatures, it has to be excited by multiple photons or by energetic particles. Therefore, the processes that are elaborated on in this work mainly apply to PAHs in UV-intense regions, where they are excited by multiple photons, or in hostile environments where they are collisionally excited by energetic particles. Also, this is further complicated by the fact that coronene should be fully dehydrogenated in PDRs (see Andrews et al. 2016) and even in the diffuse ISM (see Montillaud et al. 2013).
The 3.3 μm PAH emission band, which is commonly seen even in UV-intense reflection nebulae, planetary nebulae, and HII regions (Tielens 2008), arises predominantly from small PAHs of ∼20–30 carbon atoms (see Fig. 7 of Draine & Li 2007). Compact, pericondensed PAH molecules, such as coronene, ovalene, and circumcoronene, are exposed to the intense UV radiation in these regions and they are more likely to survive than catacondensed PAHs with an open, irregular structure. Therefore, in this work we consider coronene as a case study.
More recently, Andrews et al. (2016) modeled the physical and chemical processes of coronene, circumcoronene, and circumcircumcoronene in the northwest photodissociation region (PDR) of the reflection nebula NGC 7023 where the UV starlight intensity is ∼2600 times that of the local ISM. They found that coronene would be fully dehydrogenated in the NGC 7023 PDR. Montillaud et al. (2013) find that coronene would be fully dehydrogenated even in the diffuse ISM.
Acknowledgments
We thank the referee for his/her vey helpful comments and suggestions. This work is supported by the Swedish Research Council (Contract No. 2015-06501). The calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N). AL is supported in part by NASA 80NSSC19K0572 and NSF AST-1816411.
References
- Allamandola, L., Tielens, A. G. G. M., & Barker, J. 1985, ApJ, 290, L25 [NASA ADS] [CrossRef] [Google Scholar]
- Allamandola, L., Tielens, A. G. G. M., & Barker, J. 1989, ApJS, 71, 733 [NASA ADS] [CrossRef] [Google Scholar]
- Andrews, H., Candian, A., & Tielens, A. 2016, A&A, 595, A23 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bauschlicher, Jr., C. W. 1998, Chem. Phys., 233, 29 [Google Scholar]
- Bernstein, M. P., Sandford, S. A., & Allamandola, L. J. 1996, ApJ, 472, L127 [CrossRef] [Google Scholar]
- Bernstein, M. P., Sandford, S. A., Allamandola, L. J., et al. 1999, Science, 283, 1135 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Bernstein, M. P., Elsila, J. E., Dworkin, J. P., et al. 2002, ApJ, 576, 1115 [NASA ADS] [CrossRef] [Google Scholar]
- Burcl, R., Handy, N. C., & Carter, S. 2003, Spectrochim. Acta Part A: Mol. Biomol. Spectrosc., 59, 1881 [NASA ADS] [CrossRef] [Google Scholar]
- Castellanos, P., Candian, A., Zhen, J., Linnartz, H., & Tielens, A. G. G. M. 2018, A&A, 616, A1 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Chen, T. 2018, ApJS, 238, 18 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T. 2019, A&A, 622, A152 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Chen, T., & Luo, Y. 2019, MNRAS, 486, 1875 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T., Gatchell, M., Stockett, M. H., et al. 2014, J. Chem. Phys., 140, 224306 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Chen, T., Gatchell, M., Stockett, M. H., et al. 2015, J. Chem. Phys., 142, 144305 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T., Zhen, J., Wang, Y., Linnartz, H., & Tielens, A. G. G. M. 2018, Chem. Phys. Lett., 692, 298 [NASA ADS] [CrossRef] [Google Scholar]
- de Haas, A. J., Oomens, J., & Bouwman, J. 2017, Phys. Chem. Chem. Phys., 19, 2974 [NASA ADS] [CrossRef] [Google Scholar]
- Delaunay, R., Gatchell, M., Rousseau, P., et al. 2015, J. Phys. Chem. Lett., 6, 1536 [CrossRef] [PubMed] [Google Scholar]
- Draine, B. T., & Li, A. 2001, ApJ, 551, 807 [NASA ADS] [CrossRef] [Google Scholar]
- Draine, B. T., & Li, A. 2007, ApJ, 657, 810 [NASA ADS] [CrossRef] [Google Scholar]
- Gatchell, M., Stockett, M. H., de Ruette, N., et al. 2015, Phys. Rev. A, 92, 050702 [NASA ADS] [CrossRef] [Google Scholar]
- Gibb, E. L., Whittet, D., Schutte, W. A., et al. 2000, ApJ, 536, 347 [NASA ADS] [CrossRef] [Google Scholar]
- Hudgins, D. M., Bauschlicher, Jr., C. W., & Allamandola, L. 2005, ApJ, 632, 316 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Krasnokutski, S. A., Huisken, F., Jäger, C., & Henning, T. 2017, ApJ, 836, 32 [NASA ADS] [CrossRef] [Google Scholar]
- Leger, A., & Puget, J. 1984, A&A, 137, L5 [NASA ADS] [Google Scholar]
- Li, A., & Draine, B. T. 2002, ApJ, 572, 232 [NASA ADS] [CrossRef] [Google Scholar]
- Malloci, G., Mulas, G., Cecchi-Pestellini, C., & Joblin, C. 2008, A&A, 489, 1183 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Martín-Sómer, A., Yáñez, M., Hase, W. L., Gaigeot, M.-P., & Spezia, R. 2016, J. Chem. Theory Comput., 12, 974 [CrossRef] [Google Scholar]
- Martyna, G. J., Klein, M. L., & Tuckerman, M. 1992, J. Chem. Phys., 97, 2635 [NASA ADS] [CrossRef] [Google Scholar]
- Mattioda, A. L., Rutter, L., Parkhill, J., et al. 2008, ApJ, 680, 1243 [NASA ADS] [CrossRef] [Google Scholar]
- Micelotta, E., Jones, A., & Tielens, A. 2010, A&A, 510, A36 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Montillaud, J., Joblin, C., & Toublanc, D. 2013, A&A, 552, A15 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Nosé, S. 1984a, Mol. Phys., 52, 255 [NASA ADS] [CrossRef] [MathSciNet] [Google Scholar]
- Nosé, S. 1984b, J. Chem. Phys., 81, 511 [NASA ADS] [CrossRef] [Google Scholar]
- Paris, C., Alcamí, M., Martín, F., & Díaz-Tendero, S. 2014, J. Chem. Phys., 140, 204307 [NASA ADS] [CrossRef] [Google Scholar]
- Rapacioli, M., Cazaux, S., Foley, N., et al. 2018, Phys. Chem. Chem. Phys., 20, 22427 [NASA ADS] [CrossRef] [Google Scholar]
- Sandford, S. A., & Bernstein, M. P. 2013, ApJS, 205, 8 [NASA ADS] [CrossRef] [Google Scholar]
- Seitz, F., Zettergren, H., Rousseau, P., et al. 2013, J. Chem. Phys., 139, 034309 [NASA ADS] [CrossRef] [Google Scholar]
- Simon, A., Champeaux, J.-P., Rapacioli, M., et al. 2018, Theoret. Chem. Acc., 137, 106 [CrossRef] [Google Scholar]
- Stewart, J. J. 1989, J. Comput. Chem., 10, 209 [CrossRef] [Google Scholar]
- Stockett, M. H., Gatchell, M., Alexander, J. D., et al. 2014a, Phys. Chem. Chem. Phys., 16, 21980 [CrossRef] [PubMed] [Google Scholar]
- Stockett, M. H., Zettergren, H., Adoui, L., et al. 2014b, Phys. Rev. A, 89, 032701 [NASA ADS] [CrossRef] [Google Scholar]
- Stockett, M. H., Gatchell, M., de Ruette, N., et al. 2015, Int. J. Mass Spectrom., 392, 58 [CrossRef] [Google Scholar]
- Thrower, J., Jørgensen, B., Friis, E. E., et al. 2012, ApJ, 752, 3 [NASA ADS] [CrossRef] [Google Scholar]
- Tielens, A. G. G. M. 2008, ARA&A, 46, 289 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Tielens, A. G. G. M., Seab, C., Hollenbach, D., & McKee, C. F. 1987, ApJ, 319, L109 [NASA ADS] [CrossRef] [Google Scholar]
- Trinquier, G., Simon, A., Rapacioli, M., & Gadéa, F. X. 2017, Mol. Astrophys., 7, 37 [NASA ADS] [CrossRef] [Google Scholar]
- VandeVondele, J., Krack, M., Mohamed, F., et al. 2005, Comput. Phys. Commun., 167, 103 [NASA ADS] [CrossRef] [Google Scholar]
- West, B., Useli-Bacchitta, F., Sabbah, H., et al. 2014, J. Phys. Chem. A, 118, 7824 [CrossRef] [PubMed] [Google Scholar]
- Yang, X. J., Glaser, R., Li, A., & Zhong, J. X. 2017, New Astron. Rev., 77, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Zettergren, H., Rousseau, P., Wang, Y., et al. 2013, Phys. Rev. Lett., 110, 185501 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Zhen, J., Chen, T., & Tielens, A. G. G. M. 2018, ApJ, 863, 128 [NASA ADS] [CrossRef] [Google Scholar]
All Figures
|
Fig. 1. Structures of coronene and its seven derivatives studied in this work. The names and chemical formulae are shown beneath each molecule. |
| In the text | |
|
Fig. 2. Harmonic and anharmonic potentials. Harmonic potentials can be described by a parabolic function, which is a symmetric well, that closes on both sides. A molecule does not break up with such a harmonic potential. Anharmonic potential is an asymmetric and open well. At certain vibrational states, a molecule dissociates. |
| In the text | |
|
Fig. 3. Temperature T for coronene with vibrational energy E, calculated from its vibrational mode spectrum (see Draine & Li 2001). For coronene to attain a vibrational temperature of T ≳ 2200, 3000, and 4000 K, its vibrational energy E has to exceed ∼13.2, 20.0, and 28.5 eV, respectively. This can only be achieved for PAHs excited by multiple photons or energetic particles. |
| In the text | |
|
Fig. 4. Mass spectrum of coronene following collisions with 11.25 keV He+. The highest peaks correspond to the single and double ionizations of coronene without fragmentation on the experimental time scale of ∼10 μs. The left and right zoom-ins respectively show the intensity distributions for the losses of different numbers, n, of H atoms from the coronene dications and cations. The loss of an even number of H atoms is more favorable than the loss of an odd number of H atoms. Other peaks are due to the fragmentation of the carbon skeleton in one or several steps. See Chen et al. (2015) for details. |
| In the text | |
|
Fig. 5. Fragmentation of 1-hydrocoronene (top) and O-substituted coronene (bottom) at 3000 K. The orange text in the cells indicates the fragment(s) which are located far away from the maternal molecule. |
| In the text | |
|
Fig. 6. Fragmentation of (from top to bottom) coronene, 1-methylcoronene, coronene with a H-loss, nitrogen-substituted coronene, 1-carbonylcoronene, and 1-methoxycoronene. The simulations were performed at 4000 K. The orange text(s) in the cells indicate the fragment(s) which are located far away from the maternal molecule. |
| In the text | |
|
Fig. 7. Hydrogen roaming on C22H10 fragment produced by ejection of HCN from nitrogen-substituted coronene. |
| In the text | |
|
Fig. 8. Mass spectrum of bisanthenequinone cation (Bq+) following irradiation by laser. The three major peaks represent Bq+, [Bq-CO]+, and [Bq-2CO]+, respectively (from right to left). The optimized structures of the molecules are shown beside each peak. See Chen et al. (2018) for details. |
| In the text | |
|
Fig. 9. Formation of pentagons in nitrogen-substituted coronene. |
| In the text | |
|
Fig. 10. Formation of heptagons in 1-methoxycoronene. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.