| Issue |
A&A
Volume 622, February 2019
|
|
|---|---|---|
| Article Number | A152 | |
| Number of page(s) | 4 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/201834518 | |
| Published online | 12 February 2019 | |
Temperature effects on anharmonic infrared spectra of large compact polycyclic aromatic hydrocarbons
1
Leiden University, Leiden Observatory, Niels Bohrweg 2, 2333 CA Leiden, The Netherlands
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2
School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Theoretical Chemistry & Biology, Royal Institute of Technology, 10691 Stockholm, Sweden
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
26
October
2018
Accepted:
14
December
2018
Abstract
Aims. Large compact polycyclic aromatic hydrocarbon molecules (PAHs) present special interest in the astrochemical community. A key issue in analyses of large PAHs is understanding the effect that temperature and anharmonicity have on different vibrational bands, and thus interpreting the infrared (IR) spectra for molecules under various conditions.
Methods. Because of the huge amount of interactions/resonances in large PAHs, no anharmonic IR spectrum can be produced with static/time-independent ab initio method, especially for the molecules with D6h symmetry, e.g., coronene and circumcoronene. In this work, we performed molecular dynamics simulations to generate anharmonic IR spectra of coronene and circumcoronene.
Results. The method is validated for small PAHs, i.e., naphthalene and pyrene. We find that the semiempirical method PM3 produces accurate band positions with an error <5 cm−1. Furthermore, we calculate the spectra at multiple temperatures and find a clear trend toward band shifting and broadening.
Key words: astrochemistry / techniques: spectroscopic / line: profiles / molecular data / ISM: lines and bands / methods: laboratory: molecular
© ESO 2019
1. Introduction
It is commonly accepted that polycyclic aromatic hydrocarbon molecules (PAHs) are the carriers of discrete emission features at 3.3, 6.2, 7.7, 8.6, 11.2, and 12.7 μm bands in the infrared (IR) spectra of a variety of astrophysical environments from young stellar objects, to planetary and reflection nebulae, to the diffuse interstellar medium, and even in the emission from entire galaxies (Tielens 2008). In order to identify relevant molecular structures and characterize the physical and chemical conditions of PAHs in various astrophysical environments, experimentalists and theoreticians have been working together closely to study the IR spectra of PAHs.
From the theoretical point of view, IR vibrational spectra can be calculated using several methods. Previous computations were performed using a static/time-independent method within the framework of density functional theory (DFT), in which the double harmonic approximation is used for vibrational IR intensities. A scaling factor has to be implemented to downscale the calculated harmonic frequencies to correct the anharmonic effects or match with matrix-isolation experimental features (Langhoff 1996). As a first-order approximation, this approach has been successful enough in roughly reproducing the overall IR spectrum of certain individual PAHs, but serious problems persist when comparing this with high-resolution experiments (Mackie et al. 2015). Specifically, the IR features in the C-H stretching region are poorly modeled using harmonic level analysis, owing to the effects of mode couplings and resonances that are not described at the harmonic level (Mackie et al. 2015).
Second-order vibrational perturbations theory (VPT2) produce accurate anharmonic spectra, which compare well with experimental spectra (Chen 2018). In the VPT2 method, IR intensity can be redistributed among fundamentals, combination bands, and overtones due to resonances. A recent study shows that the standard VPT2 contains caveats with resonances; for example, the singularities that occur in resonant calculations of intensities cannot be handled correctly and this often leads to the overestimation of the intensity of certain combination bands on the spectrum. Therefore additional corrections are necessary (Mackie et al. 2015). In addition, VPT2 suffers from low efficiency of calculation and only works for small molecules (less than 24 C-atoms). Moreover, highly symmetric molecules with symmetry of D6h, for example, coronene and circumcoronene, cannot be calculated with this method. However, such species are actually expected to be highly abundant in space given their high stability (Bauschlicher et al. 2008). Circumcoronene has special interest for astronomers, as the aromatic infrared bands are believed to be carried by PAHs with more than 50 carbon atoms (Allamandola et al. 1989; Li & Draine 2001; Tielens 2008).
It has been proved that molecular dynamics (MD) can be an alternative method of VPT2 in producing anharmonic IR spectra for isolated and aqueous forms of molecules (Thicoipe et al. 2014). Simulations of MD provide the IR spectrum upon Fourier transformation of the electric dipole time-autocorrelation function. This rather general approach is most conveniently applied when the electronic ground-state potential energy surface is known beforehand and can be parametrized with force fields (Schultheis et al. 2008) or semiempirical models (Van-Oanh et al. 2002), making it particularly suitable for relatively large systems.
Alternatively, the energy surfaces can be estimated either with the Car-Parrinello molecular dynamics (CPMD; Kumar & Marx 2006 or Born-Oppenheimer molecular dynamics (BOMD; Estácio & Cabral 2008). As ab initio methods, both approaches include the effect of the electrons in the calculation of energy and forces for the classical motion of the nuclei. However, BOMD treats the electronic structure problem within the time-independent Schrödinger equation, while CPMD explicitly includes the electrons as active degrees of freedom, via (fictitious) dynamical variables. In principle, the MD simulations contain anharmonicity, resonances, combination bands ro-vibrational couplings, and temperature effects intrinsically (Horníček et al. 2007; Van-Oanh et al. 2012; Thomas et al. 2013). However, for ab initio calculations, i.e., no assumption of potential energy surface is made in advance, MD suffers from low efficiency of calculation, especially for high level theory (e.g., DFT) with a reasonable basis set to describe the vibrational features (e.g., 6-31G* or larger). To overcome this issue, in this work we combine MD with a semiempirical method to test the accuracy in producing anharmonic IR spectra of small PAHs, and apply this approach on larger compact PAHs, which cannot be obtained by other methods so far.
2. Computational details
The CP2K program with semiempirical methods of PM3 (Stewart 1989) is utilized in this work. To maintain a temperature, the Nosé-Hoover chain thermostat (Nosé 1984a,b; Martyna et al. 1992) is chosen. The structure of the molecules are optimized with PM3 before the MD simulations. In order to reach the thermal equilibration state at a give temperature, the canonical ensembles (NVT) are performed, with the optimized structure as the starting point of the NVT calculations. For generating the IR spectra, microcanonical ensemble (NVE) are followed, which provide the dipole moments at each MD step. The actual spectra are obtained by Fourier transforms of the correlation function of the dipole moments,
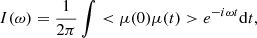 (1)
(1)
where I (ω) is the intensity of the vibrational frequency ω, and μ(t) represents the dipole moment of the molecule at time step t. In this work, the integration is performed for 50 ps with a time step of 0.5 fs to produce the vibrational spectra.
3. Results and discussion
Figure 1 shows the molecules studied in this work. Naphthalene and pyrene are chosen, because their experimental gas-phase IR spectra are available in the NIST Chemistry WebBook database (Linstrom & Mallard 2001), and it is computationally inexpensive to calculate such molecules. Therefore, they are used as a reference to validate the accuracy of the methods. Coronene and circumcoronene have neither experimental spectra in the NIST database (Linstrom & Mallard 2001) nor anharmonic IR spectra in the literature. Figure 2 shows the overall spectra, i.e., 500–3500 cm−1, of naphthalene and pyrene calculated with PM3 in comparison with the experimental gas-phase IR spectra downloaded from NIST Chemistry WebBook (Linstrom & Mallard 2001). The calculated spectra are natural, i.e., no empirical scaling factors are applied people used to do for the harmonic spectra obtained from a static quantum chemical calculation, which has been widely used in the astrophysical literature (Langhoff 1996; Bauschlicher et al. 2018). Regarding the MD simulations, for thermal equilibration, the canonical ensemble are performed for 250 ps with a time step of 0.5 fs. The temperature is set to 300 K for all the simulations and the Nosé-Hoover chain thermostat (Nosé 1984a,b; Martyna et al. 1992) with a velocity rescaling every 100 steps is used. After that, the production simulations (to produce the vibrational spectra) with microcanonical ensemble were performed for 50 ps with the same time step of 0.5 fs.
 |
Fig. 1. Structures of molecules studied in this work. Naphthalene and pyrene belong to point group D2h. Coronene and circumcoronene possess highly symmetric structure with point group D6h. |
 |
Fig. 2. Experimental and calculated gas-phase IR spectra of naphthalene and pyrene at 300 K. The top black curves represent the experimental spectra. The bottom red curves are calculated spectra using semiempirical method of PM3. The insets zoom in the region (1500–2000 cm−1) where combination bands dominate. |
One can see from Fig. 2 that the MD simulations with PM3 produce rather accurate band positions. The quantitative measurements of the C-H stretching region show that the band position of the highest peak for pyrene is ∼3050 cm−1 and PM3 gives ∼3053 cm−1, which corresponds to an error of 3 cm−1. For naphthalene, the highest peak is ∼3064 cm−1, PM3 provides ∼3060 cm−1, which corresponds to an error of 4 cm−1. The insets of Fig. 2 show the region of combination bands (1500–2000 cm−1), where anharmonicity and resonances dominate. The bands in such region are absent on the harmonic spectra (Chen 2018). We notice that the calculated spectra cannot describe the relative intensities as accurate as the band positions. This might relate to the size of the molecules, i.e., small molecules possess fewer degrees of freedom to redistribute the internal energy evenly. The larger the molecule, the more evenly the internal energy is redistributed, and the more accurate relative intensities is described.
Using PM3, we calculate the anharmonic IR spectra of neutral coronene and circumcoronene. There is no experimental spectra available for these two molecules, and the traditional time-independent methods cannot be used to compute the anharmonic spectra of such compact (D6h) molecules (Mackie et al. 2016). Figure 3 shows the calculated anharmonic spectra of coronene and circumcoronene. The harmonic spectra obtained from NASA Ames PAH Database (Bauschlicher et al. 2018) are present as a reference of the comparison. In order to correct the anharmonic effects, the spectra from the database are divided to three ranges: 1.) C-H stretching bands (> 2500 cm−1; < 4 μm), 2.) bands between 2500 and 1111.1 cm−1 (i.e., between 4 and 9 μm), and 3.) bands between 1111.1 and 0 cm−1 (i.e., > 9 μm); for each region a different scaling factor is multiplied. In addition, one or more experimental spectra are used to scale the harmonic DFT frequencies to better reproduce the experimental fundamentals (Bauschlicher et al. 2018).
 |
Fig. 3. Comparison between harmonic (top panels) and anharmonic (bottom panels) IR spectra of coronene (left panels) and circumcoronene (right panels). The harmonic spectra are downloaded from NASA Ames PAH Database, and multiple scaling factors are multiplied to correct the anharmonic effects (Bauschlicher et al. 2018). The anharmonic spectra are calculated at 300 K with semiempirical method of PM3. The insets show the region of combination bands, where anharmonicity and resonances dominate. |
The anharmonic spectra are calculated at 300 K with the semiempirical method of PM3. By comparing these two results, we can see that there are slight mismatches between the corrected harmonic spectra and the anharmonic spectra calculated using MD simulations. For instance, the C-H stretching modes for coronene is ∼3044 cm−1 (measured on the spectrum at 300 K from the MD simulation), while the corresponding value on the Ames spectrum is ∼3069 cm−1. This error (25 cm−1) is much larger than the error between the experimental spectra and the MD simulations with the semiempirical method of PM3 (see Fig. 2). A similar error in the C-H stretching region (∼21 cm−1) can also be found for circumcoronene. Based on the comparisons between experimental spectra and the calculated spectra for naphthalene and pyrene, we may infer that the scaling factors used by the Ames Database could be improved. The spectra produced by MD simulations can be used as a reference to calibrate the factors.
The PAH molecules are excited via the absorption of visible/UV photons and undergo internal conversion, in which they relax to the electronic ground state. Simultaneously, the energy is redistributed among the vibrational degree of freedom and cools down through emitting IR photons (Leger & Puget 1984; Li & Draine 2002; Mattioda et al. 2005a,b). The corresponding spectra can be severely distorted and broadened because of the temperature effects (Chen et al. 2018). Therefore for a comprehensive study of the spectral features of intersteller PAHs, it is crucial to take the temperature effects into account. To investigate the temperature effects, we run MD simulations at multiple temperatures. Four temperatures are tested: i.e., 300 K, 500 K, 700 K, and 900 K. For each temperature, the canonical ensemble calculations are performed for 250 ps for thermal equilibrations. Subsequently, the microcanonical ensemble calculations are followed for 50 ps for producing the IR spectra. Figure 4 shows the C-H stretching region of the calculated anharmonic spectra of coronene and circumcoronene at various temperatures. A clear trend of band shifting and expanding as the temperature increase can be observed. The bands systematically shift to the low-wavenumber side. However, cicumcoronene clearly shifts less than coronene; for example, for the highest peak, cicumcoronene shifts from ∼3050 cm−1 (at 300 K) to ∼3032 cm−1 (at 900 K) and therefore the shift is 18 cm−1, while coronene shifts from ∼3044 cm−1 (at 300 K) to ∼3007 cm−1 (at 900 K), which corresponds to a shift of 37 cm−1. In a follow-up paper, we will investigate the scaling law of band shifting as a function of the molecular sizes.
 |
Fig. 4. C-H stretching region of the calculated anharmonic spectra of coronene and circumcoronene at 300 K, 500 K, 700 K, and 900 K. |
4. Conclusions
In this work, we apply the molecular dynamics simulations in combination with semiempirical methods on the study of anharmonicity and temperature effects of large compact PAHs. Four molecules are investigated, naphthalene, pyrene, coronene, and circumcoronene. Naphthalene and pyrene are selected to validate the accuracy of the method. For coronene and circumcoronene, the harmonic spectra downloaded from the NASA Ames PAH Database, are used as a comparitive reference. Based on above results and discussion the general conclusions of this work are following:
-
The semiempirical method of PM3 provides accurate band positions in comparison to the experimental spectra. An error of <5 cm−1 is obtained for the C-H stretching region of naphthalene and pyrene.
-
The combination bands at 1500–2000 cm−1 region (sign of anharmonicity and resonances) can be observed on the calculated spectra, which are absent on the harmonic spectra, for example, the spectra in the NASA Ames PAH Database.
-
The MD simulations describe more accurate band positions than relative intensities, especially for small PAHs, which might be attributed to the fact that small PAHs possess fewer degrees of freedom to distribute the internal energy evenly.
-
A slight discrepancy (∼20 cm−1) between the calculated and Ames harmonic spectra can be noticed, which might because of inaccurate scaling factors in the Ames spectra.
-
The spectra at different temperatures show clear trends of band shifting and broadening. Regarding the band shifting, coronene shifts more to the low-wavenumber side than circumcoronene.
Acknowledgments
This work is supported by the Swedish Research Council (Contract No. 2015-06501). The calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N).I would like to thank Joshua P. Layfield, Timothy J. Lee, and Alexander G. G. M. Tielens for careful reading of earlier versions of this manuscript and sharing their insights on PAHs.
References
- Allamandola, L., Tielens, A., & Barker, J. 1989, ApJS, 71, 733 [NASA ADS] [CrossRef] [Google Scholar]
- Bauschlicher, Jr., C. W., Peeters, E., & Allamandola, L. J. 2008, ApJ, 678, 316 [NASA ADS] [CrossRef] [Google Scholar]
- Bauschlicher, Jr., C. W., Ricca, A., Boersma, C., & Allamandola, L. 2018, ApJS, 234, 32 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Chen, T. 2018, ApJS, 238, 18 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T., Mackie, C., Candian, A., Lee, T. J., & Tielens, A. G. G. M. 2018, A&A, 618, A49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Estácio, S. G., & Cabral, B. C. 2008, Chem. Phys. Lett., 456, 170 [NASA ADS] [CrossRef] [Google Scholar]
- Horníček, J., Kaprálová, P., & Bouř, P. 2007, J. Chem. Phys., 127, 084502 [NASA ADS] [CrossRef] [Google Scholar]
- Kumar, P., & Marx, D. 2006, PCCP, 8, 573 [NASA ADS] [CrossRef] [Google Scholar]
- Langhoff, S. R. 1996, J. Phys. Chem., 100, 2819 [CrossRef] [Google Scholar]
- Leger, A., & Puget, J. 1984, A&A, 137, L5 [NASA ADS] [Google Scholar]
- Li, A., & Draine, B. 2001, ApJ, 554, 778 [Google Scholar]
- Li, A., & Draine, B. 2002, ApJ, 572, 232 [NASA ADS] [CrossRef] [Google Scholar]
- Linstrom, P. J., & Mallard, W. G. 2001, J. Chem. Eng. Data, 46, 1059 [Google Scholar]
- Mackie, C. J., Candian, A., Huang, X., et al. 2015, J. Chem. Phys., 143, 224314 [NASA ADS] [CrossRef] [Google Scholar]
- Mackie, C. J., Candian, A., Huang, X., et al. 2016, J. Chem. Phys., 145, 084313 [NASA ADS] [CrossRef] [Google Scholar]
- Martyna, G. J., Klein, M. L., & Tuckerman, M. 1992, J. Chem. Phys., 97, 2635 [NASA ADS] [CrossRef] [Google Scholar]
- Mattioda, A., Allamandola, L., & Hudgins, D. 2005a, ApJ, 629, 1183 [NASA ADS] [CrossRef] [Google Scholar]
- Mattioda, A., Hudgins, D., & Allamandola, L. 2005b, ApJ, 629, 1188 [NASA ADS] [CrossRef] [Google Scholar]
- Nosé, S. 1984a, J. Chem. Phys., 81, 511 [NASA ADS] [CrossRef] [Google Scholar]
- Nosé, S. 1984b, Mol. Phys., 52, 255 [NASA ADS] [CrossRef] [MathSciNet] [Google Scholar]
- Schultheis, V., Reichold, R., Schropp, B., & Tavan, P. 2008, J. Phys. Chem. B, 112, 12217 [CrossRef] [Google Scholar]
- Stewart, J. J. 1989, J. Comput. Chem., 10, 209 [CrossRef] [Google Scholar]
- Thicoipe, S., Carbonniere, P., & Pouchan, C. 2014, Chem. Phys. Lett., 591, 243 [NASA ADS] [CrossRef] [Google Scholar]
- Thomas, M., Brehm, M., Fligg, R., Vöhringer, P., & Kirchner, B. 2013, PCCP, 15, 6608 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Tielens, A. G. G. M. 2008, ARA&A, 46, 289 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Van-Oanh, N.-T., Parneix, P., & Bréchignac, P. 2002, J. Phys. Chem. A, 106, 10144 [CrossRef] [Google Scholar]
- Van-Oanh, N.-T., Falvo, C., Calvo, F., et al. 2012, PCCP, 14, 2381 [NASA ADS] [CrossRef] [Google Scholar]
All Figures
|
Fig. 1. Structures of molecules studied in this work. Naphthalene and pyrene belong to point group D2h. Coronene and circumcoronene possess highly symmetric structure with point group D6h. |
| In the text | |
|
Fig. 2. Experimental and calculated gas-phase IR spectra of naphthalene and pyrene at 300 K. The top black curves represent the experimental spectra. The bottom red curves are calculated spectra using semiempirical method of PM3. The insets zoom in the region (1500–2000 cm−1) where combination bands dominate. |
| In the text | |
|
Fig. 3. Comparison between harmonic (top panels) and anharmonic (bottom panels) IR spectra of coronene (left panels) and circumcoronene (right panels). The harmonic spectra are downloaded from NASA Ames PAH Database, and multiple scaling factors are multiplied to correct the anharmonic effects (Bauschlicher et al. 2018). The anharmonic spectra are calculated at 300 K with semiempirical method of PM3. The insets show the region of combination bands, where anharmonicity and resonances dominate. |
| In the text | |
|
Fig. 4. C-H stretching region of the calculated anharmonic spectra of coronene and circumcoronene at 300 K, 500 K, 700 K, and 900 K. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.