| Issue |
A&A
Volume 581, September 2015
|
|
|---|---|---|
| Article Number | A92 | |
| Number of page(s) | 7 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201526487 | |
| Published online | 11 September 2015 | |
H2 formation via the UV photo-processing of a-C:H nano-particles
Université Paris Sud and CNRS, Institut d’Astrophysique Spatiale, UMR
8617,
91405
Orsay,
France
e-mail:
This email address is being protected from spambots. You need JavaScript enabled to view it.
Received: 7 May 2015
Accepted: 26 July 2015
Abstract
Context. The photolysis of hydrogenated amorphous carbon, a-C(:H), dust by UV photon-irradiation in the laboratory leads to the release of H2 as well as other molecules and radicals. This same process is also likely to be important in the interstellar medium.
Aims. We investigate molecule formation arising from the photo-dissociatively-driven, regenerative processing of a-C(:H) dust.
Methods. We explore the mechanism of a-C(:H) grain photolysis leading to the formation of H2 and other molecules/radicals.
Results. The rate constant for the photon-driven formation of H2 from a-C(:H) grains is estimated to be 2 × 10-17 cm3 s-1. In intense radiation fields photon-driven grain decomposition will lead to fragmentation into daughter species rather than H2 formation.
Conclusions. The cyclic re-structuring of arophatic a-C(:H) nano-particles appears to be a viable route to formation of H2 for low to moderate radiation field intensities (1 ≲ G0 ≲ 102), even when the dust is warm (T ~ 50−100 K).
Key words: dust, extinction / ISM: molecules / ISM: general
© ESO, 2015
1. Introduction
The evolution of hydrocarbon solids was considered in detail in our preceding works (Jones 1990, 2009, 2012a,b,c, 2014; Jones et al. 2013, 2014). Here we extend that work and explore the formation of molecules arising from the vacuum ultraviolet (VUV, E ~ 7−10 eV) and extreme ultraviolet (EUV, E ~ 10−13.6 eV) photon-driven processing and decompositional evolution of hydrogenated amorphous carbon particles, a-C(:H), in the interstellar medium (ISM). The designation a-C(:H) is used to indicate the entire range of hydrogen-poor (a-C) to hydrogen-rich (a-C:H) carbonaceous solids. The UV photolysis or ion irradiation a-C:H materials leads to their aromatisation, H atom loss and the in situ formation of molecular hydrogen (e.g., Smith 1984; Adel et al. 1989; Marée et al. 1996; Godard et al. 2011; Alata et al. 2014). The most recent of these experiments by Alata et al. (2014) show that the VUV (E ~ 6.8−10.5 eV, λ ~ 120−180 nm) photolysis of hydrogenated amorphous carbons is indeed a viable mechanism for the formation of H2, and also CH4, within an astrophysical framework. The photo-processing of a-C(:H) particles by VUV/EUV photons (hereafter referred to as UV photons)1 from the ambient ISRF leads to re-structuring/decomposition, H2 formation and the liberation of H2 and hydrocarbon daughter products (e.g., Smith 1984; Alata et al. 2014; Duley et al. 2015). This process could also liberate (the precursors to) species such as CCH, c-C3H2, C3H+, C4H, which have been observed in ISM regions with moderate UV radiation fields (e.g., Pety et al. 2005, 2012; Guzmán et al. 2015).
In the classical picture of H2 formation in the ISM an H atom physisorbed on a grain surface2 reacts with a second from the gas phase (Eley-Rideal mechanism) or with another on the grain surface (Langmuir-Hinshelwood mechanism) before reacting to form an H2 molecule which is then ejected into the gas phase. In their recent study Bron et al. (2014) showed that the Langmuir-Hinshelwood mechanism on small grains with fluctuating temperatures can still be an efficient route to H2 formation in unshielded photon dominated regions (PDRs). The detailed analysis of Bron et al. (2014) shows that surface reactions on small, stochastically-heated particles are therefore no barrier to H2 formation in PDRs and can provide means to form H2 where the classical mechanisms do not appear to work. Here we describe a new mechanism for H2 formation via the photolysis of stochastically-heated a-C nano-particles, which is complementary to that proposed by Bron et al. (2014). In this new scenario the H atoms involved in H2 molecule formation are chemically-bonded3 within the grains, as in the experiments of Alata et al. (2014). The mechanism appears to be viable route to molecule formation in PDRs with moderate-strength radiation fields where the grains are warm.
In this paper we propose a molecule/radical formation pathway via the UV photon-driven C−H and C−C bond dissociation within, and the associated decomposition of, a-C(:H) nano-particles, which is consistent with carbonaceous dust evolution in the ISM. The paper is organised as follows: Sect. 2 proposes a new mechanism for molecular hydrogen and daughter molecule formation, Sect.3 quantifies H2 formation in a-C(:H) solids and Sect. 4 presents the conclusions of this work.
2. A mechanism for molecule formation
 |
Fig. 1 Schematic view of part of the 3D structure of an a-C(:H) grain containing ~60 C atoms and ~40 H atoms. Some of the aliphatic, olefinic and aromatic H atoms inherent within the structure are indicated by the red, blue and purple filled circles, respectively. Radical C atom or dangling bond sites sites are shown by the black dots. See text for details. |
Experimental evidence shows that methyl groups, CH3, within a-C:H materials probably play an important role in H2 and CH4 formation during UV photolysis (e.g., Alata et al. 2014). In the diffuse ISM and PDRs it will be the small, stochastically-heated grains that dominate H2 formation because of the large surface area that they provide (Bron et al. 2014). In the Jones et al. (2013) dust model the smallest grains (a< 10 nm) are narrow band gap (Eg = 0.1 eV), H-poor, a-C nano-particles with no methyl groups apparent in their structure (Jones 2012a,b,c). Thus, in the tenuous ISM it is unlikely that methyl groups would contribute to H2 formation.
The key structural component in molecule formation from and within a-C nano-particles (a ≲ 5 nm, Jones 2012c) would appear to be the presence of the aliphatic and olefinic bridging groups, which link the aromatic domains in these arophatic structures (e.g., Jones 2012c; Micelotta et al. 2012). Figure 1 shows a schematic view of part of an a-C(:H) nano-particle where the aliphatic −CH2 −CH2 − and olefinic −CH=CH− bridges between aromatic clusters are indicated by the ellipsoidal areas A and B, respectively. A longer −C4H6 − mixed aliphatic-olefinic bridge is indicated by the dashed ellipse on the right of Fig. 1. The de-hydrogenation and dissociation of such structures could be at the heart of molecule formation within a-C(:H) grains.
H2 molecule formation in a-C(:H) is somewhat analogous to that arising from the aromatic-aliphatic transformation on the periphery of PAH molecules proposed by Rauls & Hornekær (2008) and the catalytic formation of H2 by coronene as studied experimentally by Mennella et al. (2012). However, it is here hypothesised that H2 formation occurs at the aliphatic/olefinic bridging structures that link the aromatic clusters in a-C(:H) in nano-particles (e.g., the A and B sites in Fig. 1). As Ferrari & Robertson (2004), and Hu et al. (2007a,b) point out, sp2 chain and cage-like clusters may be rather common structures in a-C:H materials. Our own work indicates aromatic sp2 clusters containing 3−8 rings in a-C nano-particles with radii ≃0.2−0.4 nm (Jones 2012c; Micelotta et al. 2012). As Fig. 1 shows the aromatic domains and the aliphatic/olefinic bridges in these arophatic-type structures are not co-planar but rather cage-like. Hence, under the effects of CH bond UV photo-dissociation the aliphatic/olefinic (A/B) bridging structures are unable to transform into aromatic structures, or increase the aromatic domain size, without compromising the cage-like structure, i.e., without breaking CC bonds. An aliphatic bridge (A) can, however, transform into an olefinic bridge (B), which is likely to be sterically-strained because of the distortions introduced by the random network structure. The olefinic bridge could then flip between olefinic (B) and radical aliphatic states (C) with dangling bonds, i.e., undergo B⇔C isomerisational flipping. Such a mechanism is also consistent with the formation of daughter hydrocarbon radicals and molecules.
For a simple, two-carbon atom aliphatic bridge, A, the underlying H2 formation process depends on an equilibrium between UV photolysis and H atom addition (e.g., Muñoz Caro et al. 2001; Mennella et al. 2001; Alata et al. 2014, see also Sect. 3). The mechanism for H2 formation proposed here relies on the aliphatic bridges being dehydrogenated, i.e.,  and H atom addition being aided by the potential for sterically-hindered, de-hydrogenated aliphatic bridges to isomerise, i.e.
and H atom addition being aided by the potential for sterically-hindered, de-hydrogenated aliphatic bridges to isomerise, i.e.  where A, B, C, etc. also refer to Fig. 1 and the • symbol is used to indicate a radical species with dangling bonds on a carbon atom that can chemisorb H atoms. As detailed above this isomerisation scheme involves an aliphatic-to-olefinic transition that cannot proceed to aromatisation in arophatic structures because of steric limitations (e.g., Jones 2012c; Micelotta et al. 2012).
where A, B, C, etc. also refer to Fig. 1 and the • symbol is used to indicate a radical species with dangling bonds on a carbon atom that can chemisorb H atoms. As detailed above this isomerisation scheme involves an aliphatic-to-olefinic transition that cannot proceed to aromatisation in arophatic structures because of steric limitations (e.g., Jones 2012c; Micelotta et al. 2012).
As implied above, and adopted in the following analysis, a UV photon is assumed to dissociate a single C−H bond releasing an H atom that can them react with a bonded H atom to form H2. In the above, and for simplicity in what follows, we show adjacent H atoms forming an H2 molecule. However, the reacting H atoms need not necessarily be adjacent because the C−H bond dissociation-released H atom can migrate through the structure and react with a more distant H atom4.
The full sequence for H2 formation and aliphatic bridging structure re-cycling can be summarised by the following schematic of the UV-catalysed, H2 formation cycle:
![Mathematical equation: \begin{eqnarray*} &&-{\rm CH}_2 - {\rm CH}_2- \ \ \ [{\rm aliphatic\ bridge}] \ \rightarrow \\ && \hspace{0.9cm} {\rm A} \\ && ({h\nu^\ast} : {\rm H}_2) \ \rightarrow \ -{\rm C}^\bullet {\rm H}-{\rm C}^\bullet{\rm H}- \leftrightarrow -{\rm CH}={\rm CH}- \\ &&\hspace{3.05cm} {\rm C} \hspace{2.25cm} {\rm B} \\ && ({\rm H}:\ ) \ \ \ \ \ \ \ \ \rightarrow \ -{\rm CH_2}-{\rm C}^\bullet{\rm H}- \\ && \hspace{2.8cm} {\rm A/C} \\ && ({\rm H}:\ ) \ \ \ \ \ \ \ \ \rightarrow \ -{\rm CH_2}-{\rm CH_2}- \ [{\rm re-generated\ aliphatic\ bridge}] \\ && \hspace{3.0cm} {\rm A} \end{eqnarray*}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq31.png) where we adopt the descriptor (reactant : product): with hν∗ and H being dehydrogenating UV photons and adsorbing H atoms, respectively, and H2 the UV-photolysis product. The label A/C is used to indicate a mixed A and C bridge. For this H2 formation cycle to operate efficiently it relies on the re-generating H atom addition time-scale being, at least, of same order of magnitude as that for the UV-photolysis step, which appears to be the case (see Sect. 3.2). However, as the radiation field intensity increases the cycle will be increasingly interrupted by the competing dehydrogenation processes
where we adopt the descriptor (reactant : product): with hν∗ and H being dehydrogenating UV photons and adsorbing H atoms, respectively, and H2 the UV-photolysis product. The label A/C is used to indicate a mixed A and C bridge. For this H2 formation cycle to operate efficiently it relies on the re-generating H atom addition time-scale being, at least, of same order of magnitude as that for the UV-photolysis step, which appears to be the case (see Sect. 3.2). However, as the radiation field intensity increases the cycle will be increasingly interrupted by the competing dehydrogenation processes
 i.e., at some point the UV-photolysed dehydrogenation becomes more important than H atom addition. In the case of extremely intense radiation fields, the UV-photolysis rate will exceed the H atom addition rate and H2 formation will no longer be possible. In this case, the above processes can lead to photo-fragmentation of the structure (e.g., Micelotta et al. 2010b,a; Bocchio et al. 2012; Jones et al. 2013) and the formation of daughter molecule/radical species, such as C
i.e., at some point the UV-photolysed dehydrogenation becomes more important than H atom addition. In the case of extremely intense radiation fields, the UV-photolysis rate will exceed the H atom addition rate and H2 formation will no longer be possible. In this case, the above processes can lead to photo-fragmentation of the structure (e.g., Micelotta et al. 2010b,a; Bocchio et al. 2012; Jones et al. 2013) and the formation of daughter molecule/radical species, such as C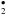 H and C2, as the particles undergo UV-photon-induced catastrophic destruction.
H and C2, as the particles undergo UV-photon-induced catastrophic destruction.
If the aliphatic/olefinic bridges are longer, as shown by the dotted ellipse in Fig. 1, i.e.,
–CH2–CH=CH-CH2– ,
–CH2–CH2–CH2– or
–CH2–CH2–CH2–CH2– ,
then a whole series of fragments, with the generic formula CnH(m≤2n), where n = 2,3,4... and m = 0 → 2n, are possible (e.g., see Jones 2012b; Duley et al. 2015). It appears that such an a-C(:H) decomposition mechanism could perhaps better explain the origin of the observed C2H, C3H, l-C3H+, c-C3H2, l-C3H2 and C4H daughter products observed in the Horsehead Nebula PDR by Pety et al. (2005, 2012) and Guzmán et al. (2015). These species would then originate from the UV photo-fragmentation of the aliphatic and olefinic structures that bind the aromatic clusters in a-C(:H) nano-particles, rather than from the photo-dissociation of free-flying precursor PAHs. In the most recent of these works Guzmán et al. (2015) show that in the Horsehead Nebula PDR the IR band emission, H2 2.12μm ro-vibrational line and the l–C3H+ emission peaks are coincident and on the leading edge of the PDR (AV< 1) just in front of the C2H and c-C3H2 emission peaks at AV ~ 1. The latter, neutral hydrocarbon species are consistent with the break up of the a-C(:H) aliphatic/olefinic bridges in the PDR and the release of aromatic clusters into the gas that are then excited on the strongly-illuminated face of the PDR.
Rather convincing evidence for an intimate mix of aliphatics with aromatics in carbonaceous nano-particles in the ISM (see the Fig. 1 schematic view), as predicted by Jones (2012c) and Jones et al. (2013), comes from the observed 3−4 μm region emission band spectra in many sources, e.g., NGC 7023 (Pilleri et al. 2015), the Orion Bar (Sloan et al. 1997; Verstraete et al. 2001), M17 SW, NGC 2023 (Verstraete et al. 2001), M 82 (Yamagishi et al. 2012) and the Seyfert 1 galaxy NGC 1097 (Kondo et al. 2012). Even in these energetic regions the 3.3 μm aromatic CH band always appears to be accompanied by a CHn aliphatic plateau in the 3.4−3.6 μm region. These observations are hard to explain within the context of PAH molecules in the strict chemical sense, i.e., polycyclic aromatics with only sp2 C atoms with peripheral H atoms. The “interstellar PAH” model therefore requires that the IR emission band band-carriers are more complex species than PAHs (e.g., Li & Draine 2012) and are probably more akin to the a-C(:H) nano-particle arophatics (Micelotta et al. 2012) that we propose are at the heart of H2 molecule formation in PDRs (see Fig. 1).
3. The molecular hydrogen formation rate
The formation of H2, associated with the aliphatic to aromatic transformation of a-C(:H) dust, should also occur in the ISM where UV photons progressively de-hydrogenate and aromatise a-C(:H) materials. This coupled dehydrogenation/aromatisation process will clearly be accompanied by a de-volatilisation due to the associated loss of small molecules (e.g., Smith 1984; Jones 2012b; Alata et al. 2014; Duley et al. 2015). Thus, the chemistry in PDRs would appear to be an ideal laboratory to search for tracers of this H2 and small molecule formation mechanism, which will depend upon the radiation field intensity rather than, simply, the gas density.
The H2 formation rate in the ISM, dn [ H2 ]/dt, is classically described by the balance between its UV photo-destruction in the gas phase and its formation by H atom recombination on the surfaces of dust particles, i.e., ![Mathematical equation: \begin{equation} \frac{{\rm d}n[{\rm H_2}]}{{\rm d}t} = -\ \beta_0 \, n_{\rm H_2} \ + \ R_{\rm H_2} \, n_{\rm p} \, n_{\rm H}, \label{eq_H2_equilibrium} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq47.png) (1)where β0 is the H2 photo-dissociation rate coefficient (5 × 10-11 s-1) in an unshielded interstellar radiation field (ISRF), RH2 is the H2 formation rate coefficient for formation via H atom sticking and recombination on dust, np is the total proton density, and nH and nH2 are the atomic and molecular hydrogen number densities, respectively. The observationally-determined H2 formation rate coefficient in the diffuse ISM, RH2 ≃ 3−4 × 10-17 cm3 s-1 (e.g., Gry et al. 2002), appears to be rather invariant, indicating a degree of uniformity in the H2 formation process.
(1)where β0 is the H2 photo-dissociation rate coefficient (5 × 10-11 s-1) in an unshielded interstellar radiation field (ISRF), RH2 is the H2 formation rate coefficient for formation via H atom sticking and recombination on dust, np is the total proton density, and nH and nH2 are the atomic and molecular hydrogen number densities, respectively. The observationally-determined H2 formation rate coefficient in the diffuse ISM, RH2 ≃ 3−4 × 10-17 cm3 s-1 (e.g., Gry et al. 2002), appears to be rather invariant, indicating a degree of uniformity in the H2 formation process.
Alata et al. (2014) recently showed that the VUV photo-processing (≈8.6 eV per photon) of a-C:H leads to bond photo-dissociation, decomposition and the formation of H2within the a-C(:H) bulk material. The H2 formation rate, RH2, is then a first order reaction with rate constant kf [ CH ] and Eq. (1) becomes ![Mathematical equation: \begin{equation} \frac{{\rm d}n[{\rm H_2}]}{{\rm d}t} = -\ \beta_0 \, n_{\rm H_2} \ + \ k_f [{\rm CH}] \, n_{\rm p} \label{eq_H2_form_rate} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq57.png) (2)and as per Alata et al. (2014), in their Sect. 4.1, both terms on the right hand side of Eq. (2), i.e., the H2 destruction and formation rates, are directly proportional to the UV photon flux5. As given by Eq. (39) in Jones (2012b), kf [ CH ] can be expressed as
(2)and as per Alata et al. (2014), in their Sect. 4.1, both terms on the right hand side of Eq. (2), i.e., the H2 destruction and formation rates, are directly proportional to the UV photon flux5. As given by Eq. (39) in Jones (2012b), kf [ CH ] can be expressed as ![Mathematical equation: \begin{equation} k_f [{\rm CH}] = \Lambda_{\rm UV,pd}(a) \ N_{\rm CH}(a,d_1) \ X_i(a) \ \ \ {\rm [\,s^{-1}]} \label{eq_H2_formation_rate} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq58.png) (3)where ΛUV,pd(a) is the UV photo-darkening6 rate (Eq. (31) Jones 2012b), Xi(a) is the grain relative abundance, NCH(a,d1) is the number of CH bonds in a grain of radius a photolysable to a depth d1 (≃20 nm)7 and therefore capable of forming H2. Equation (3) implicitly assumes that every absorbed UV photon leads to the photo-dissociation of a single CH bond and the formation of an H2 molecule. This is almost certainly not the case because there will be competition from other channels, such as photon absorption leading to grain heating or photo-electron emission. Recent experimental measurements indicate low photo-dissociation efficiencies, i.e., 0.014 (Maté 2014) and 0.025 (Alata et al. 2014) for the CH bonds in bulk a-C:H materials. Here we assume a size-dependent photo-dissociation efficiency ϵ = 2 /a [nm] for a ≥ 2 nm and ϵ = 1 for a< 1 nm (see Jones 2012b,d; Jones et al. 2014, for further details)8. Following Jones (2012b,d) the UV photo-darkening rate, and hence the H2 formation rate, is given by
(3)where ΛUV,pd(a) is the UV photo-darkening6 rate (Eq. (31) Jones 2012b), Xi(a) is the grain relative abundance, NCH(a,d1) is the number of CH bonds in a grain of radius a photolysable to a depth d1 (≃20 nm)7 and therefore capable of forming H2. Equation (3) implicitly assumes that every absorbed UV photon leads to the photo-dissociation of a single CH bond and the formation of an H2 molecule. This is almost certainly not the case because there will be competition from other channels, such as photon absorption leading to grain heating or photo-electron emission. Recent experimental measurements indicate low photo-dissociation efficiencies, i.e., 0.014 (Maté 2014) and 0.025 (Alata et al. 2014) for the CH bonds in bulk a-C:H materials. Here we assume a size-dependent photo-dissociation efficiency ϵ = 2 /a [nm] for a ≥ 2 nm and ϵ = 1 for a< 1 nm (see Jones 2012b,d; Jones et al. 2014, for further details)8. Following Jones (2012b,d) the UV photo-darkening rate, and hence the H2 formation rate, is given by ![Mathematical equation: \begin{equation} \Lambda_{\rm UV,pd}(a) = F_{\rm UV} \ \sigma_{\rm CH-diss.} \ Q_{\rm abs}(a,E) \, \epsilon\ \ \ {\rm [\,s^{-1}]}, \label{eq_UV_dehyd_rate} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq68.png) (4)where FUV ≃ 3 × 107 photons cm-2 s-1 is the CH bond-dissociating UV photon flux in the local ISM (Henry 2002) for G0 = 1, σCH−diss. ≃ 10-19 cm2 is the CH bond photo-dissociation cross-section for Ehν ≳ 10 eV (Welch & Judge 1972; Gruzdkov et al. 1994; Mennella et al. 2001; Muñoz Caro et al. 2001; Alata et al. 2014) and Qabs(a,E) is the particle absorption efficiency factor in the cross-section, i.e., σ(a,E) = πa2Qabs(a,E). The a-C(:H) particle photo-processing time-scale, as a function of grain radius and the radiation field, can be expressed as
(4)where FUV ≃ 3 × 107 photons cm-2 s-1 is the CH bond-dissociating UV photon flux in the local ISM (Henry 2002) for G0 = 1, σCH−diss. ≃ 10-19 cm2 is the CH bond photo-dissociation cross-section for Ehν ≳ 10 eV (Welch & Judge 1972; Gruzdkov et al. 1994; Mennella et al. 2001; Muñoz Caro et al. 2001; Alata et al. 2014) and Qabs(a,E) is the particle absorption efficiency factor in the cross-section, i.e., σ(a,E) = πa2Qabs(a,E). The a-C(:H) particle photo-processing time-scale, as a function of grain radius and the radiation field, can be expressed as  (5)From Eq. (2) the molecular hydrogen fraction, at equilibrium (dn [ H2 ]/dt = 0), is
(5)From Eq. (2) the molecular hydrogen fraction, at equilibrium (dn [ H2 ]/dt = 0), is ![Mathematical equation: \begin{eqnarray} \frac{n_{\rm H_2}}{n_{\rm H}} &=& \frac{k_f [{\rm CH}]}{\beta_0} \nonumber \\ &=& \left[ \frac{F_{\rm UV} \, \sigma_{\rm CH-diss.}}{\beta_0} \right] \Bigg\{ Q_{\rm abs}(a,E) \, \epsilon(a) \,N_{\rm CH}(a,d_1) \, X_i(a) \Bigg\}. \label{eq_H2_equilib_frac} \end{eqnarray}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq78.png) (6)Alata et al. (2014) derived a similar expression for this ratio (see in their Sect. 4.2) but did not include the size-dependent optical property terms for the dust.
(6)Alata et al. (2014) derived a similar expression for this ratio (see in their Sect. 4.2) but did not include the size-dependent optical property terms for the dust.
Within the framework of the proposed model, the UV photon flux is therefore key in determining the H2 formation and destruction rates, which scale with the local ISRF intensity and hardness. However, even if H2 formation via a-C:H UV photolysis is viable, it will be hampered by the limited stock of H atoms within the dust, which is determined by the carbon depletion into dust, i.e., [ C / H ] ≈ 10-4. As shown by Jones (2012b), H2 formation would be sustainable for only ≈104 yr before all CH bonds were photo-dissociated and the grains completely de-hydrogenated during aromatisation. An a-C re-hydrogenation mechanism, such as that proposed by Mennella (2008, 2010), is therefore needed to replenish the stock of CH bonds. Thus, the complete dehydrogenation/aromatisation of a-C(:H) grains must be hindered and suitably stable and numerous H atom chemisorption sites need to be preserved. These sites could be dangling bonds resulting from steric limitations on the sp3 to sp2 transformation (see Sect. 2) or due to the low-level doping of a-C(:H) with nitrogen atoms (e.g., Amir & Kalish 1991; Jones 2013, see Sect. 3.1). In this case, H2 formation would be sustainable as long as the radiation field is intense enough to photo-dissociate CH bonds but not intense enough to break the CC bonds in the aliphatic/olefinic bridging structure and hence to photo-fragment the a-C(:H) grains. Thus, this H2 formation mechanism will not work at high extinction because there are too few UV photons, nor will it work in intense radiation fields because of the rapid photo-fragmentation of the grains, i.e., their destruction via the formation and ejection of daughter molecules/radicals (e.g., Smith 1984; Pety et al. 2005; Jones 2012b; Jones et al. 2013; Alata et al. 2014, see also Sect. 3.3). These ideas appear to be in general agreement with the conclusions of Habart et al. (2004, 2011), who find enhanced H2 formation and higher than expected column densities of rotationally excited H2 in moderately-excited PDRs.
3.1. A role for nitrogen heteroatoms?
Apparently, heteroatom groups, i.e., chemical groups containing atoms other than C or H, are of low abundance or are absent from carbonaceous materials in the ISM (Pendleton & Allamandola 2002; Dartois et al. 2005). Nevertheless, N atoms were clearly detected in the comet Halley CHON carbonaceous phase (in CHON and CHN combinations, Clark et al. 1987) indicating N hetero-atoms within solar system carbonaceous dust.
Interestingly, a-C:H materials can be rather inefficiently doped with nitrogen atoms to form a-C:H:N materials (Schwan et al. 1994) and this could have some interesting consequences for the observable properties of a-C(:H) dust in the ISM and circumstellar regions (Jones 2013, 2014). Amir & Kalish (1991) find that low N-doped a-C:H materials (N concentration <1 atomic %), with an optical gaps ≃1 eV, are rich with dangling bonds9 at a concentration of 5 × 1020 cm-3. N heteroatom-induced dangling bonds could provide H atom trapping sites for the catalytic formation of H2 in N-doped arophatics.
The inclusion of N atoms into amorphous carbons to form amorphous carbon nitrides, a-CNx, has led to the classification of a number of N-bonding configurations (e.g., Ferrari & Robertson 2004). In their experimental study Dartois et al. (2005) found that the nitrogen atoms in their laboratory interstellar a-C:H analogues tend to be found in structure-bridging groups rather than in aromatic clusters. However, the similar frequencies for CC and CN modes can make the interpretation of the skeletal modes difficult (e.g., Ferrari & Robertson 2004).
 |
Fig. 2 Canonical interstellar H2 formation rate constant (grey horizontal band, Gry et al. 2002) compared to that predicted for H2 formation via CH bond UV photo-dissociation from a-C:H/a-C grains, kf[CH]/nH = 2.0 × 10-17 cm3 s-1 shown at a = 0.5 nm (squares with error bars). The green green square also includes H2 formation in the a-C mantles on the large a-SilFe grains into the calculation, yielding kf[CH]/nH = 4.0 × 10-17 cm3 s-1. The small squares show the relative contributions to the total H2 formation rate as a function of particle radius. The H atom sticking rate constant, for sH = 1, is shown for the small a-C (thin grey), large a-C:H/a-C (thick grey) and a-Sil/a-C (green) grain distributions and for gas temperatures of 50, 100 and 200 K (short-dashed, solid and long-dashed, respectively). |
3.2. The dust model and H2 formation
In this work we adopt the dust compositions and size distributions from the Jones et al. (2013), Köhler et al. (2014) dust model, which is specifically-tuned to the evolution of the optical and thermal properties of a-C(:H) grains in the ISM (i.e., using the optEC(s) and optEC(s)(a) data from Jones 2012b,c,d,e). The Jones et al. (2013) model consists of a power-law distribution of small a-C grains, with a bulk material band gap Eg = 0.1 eV, and a log-normal distribution of a-C:H/a-C (core/mantle) grains, with Eg = 2.5 eV and 0.1 eV, respectively. These hydrocarbon populations are complemented by a log-normal distribution of large amorphous forsterite- and pyroxene-type silicate grains, containing iron and iron sulphide nano-particle inclusions, and with a 5 nm thick coating of a-C (a-SilFe/a-C, Jones et al. 2013; Köhler et al. 2014). The model, strongly-constrained by laboratory-data, is able to explain the observed variations in the IR-FUV extinction, the 217 nm UV bump, the IR absorption and emission bands and the IR-mm dust thermal emission.
The H atom collision and sticking rate coefficient for a grain of radius a and relative abundance Xi(a) is given by  (7)where Tk is the gas kinetic temperature and sH is the H atom sticking efficiency. Figure 2 shows this rate coefficient, RH/nH, assuming sH = 1, for gas temperatures of 50, 100 and 200 K.
(7)where Tk is the gas kinetic temperature and sH is the H atom sticking efficiency. Figure 2 shows this rate coefficient, RH/nH, assuming sH = 1, for gas temperatures of 50, 100 and 200 K.
The nH-normalised, UV photon-induced, H2 formation rate coefficients, kf [ CH ] /nH, integrated over the small and large a-C(:H) grain size distributions are indicated in Fig. 2 by the large squares with error bars. These data give an good fit to the observationally-derived H2 formation rate coefficient in the diffuse ISM, i.e., RH2 ≃ 3−4 × 10-17 cm3 s-1 (Gry et al. 2002), which is indicated by the horizontal grey band.
The size-dependent a-C(:H) particle photo-darkening or CH bond photo-dissociation time-scale in the diffuse ISM was recently estimated by Jones et al. (2014). They found that the a-C(:H) photo-processing time-scale can be analytically expressed as a function of the grain radius, a, in nm; ![Mathematical equation: \begin{equation} \tau_{\rm UV,pd}(a) = \frac{10^4}{G_0} \bigg\{ 2.7 + \frac{6.5 }{(a\,[{\rm nm}])^{1.4}} + 0.04 \, (a\,[{\rm nm}])^{1.3} \bigg\} \ \ {\rm [yr]}, \label{eq_pd_timescale} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq102.png) (8)which implies a minimum processing time-scale, of the order of (4 × 104/G0) yr, for particles with a ~ 3−10 nm and processing time-scales
(8)which implies a minimum processing time-scale, of the order of (4 × 104/G0) yr, for particles with a ~ 3−10 nm and processing time-scales  (105/G0) yr for the smallest and largest grains in the assumed a-C(:H) particle size distribution (Jones et al. 2014). Here we assume the particle size-dependent, photo-darkening efficiency factor, ϵ(a) given above (see Sect. 3). For a-C(:H) grains such a size-dependent CH bond photo-dissociation efficiency appears to be a physically realistic because with increasing particle size other channels will compete with CH photo-dissociation, e.g., UV photon absorption leading to thermal excitation and/or fluorescence. However, the proposed H2 formation mechanism is only viable if the H atom sticking rate is higher than the CH photo-dissociation rate and Fig. 2 shows that this is indeed the case. We find an H2 formation rate constant of 2.0 × 10-17 cm3 s-1 for a-C:H/a-C grains and 4.0 × 10-17 cm3 s-1 if the a-C coated amorphous silicate grains, a-Sil/a-C, are also included. The error bar on the square data points in Fig. 2 conservatively reflects a factor of three uncertainty in the dust optical properties and a factor of ten uncertainty in ϵ.
(105/G0) yr for the smallest and largest grains in the assumed a-C(:H) particle size distribution (Jones et al. 2014). Here we assume the particle size-dependent, photo-darkening efficiency factor, ϵ(a) given above (see Sect. 3). For a-C(:H) grains such a size-dependent CH bond photo-dissociation efficiency appears to be a physically realistic because with increasing particle size other channels will compete with CH photo-dissociation, e.g., UV photon absorption leading to thermal excitation and/or fluorescence. However, the proposed H2 formation mechanism is only viable if the H atom sticking rate is higher than the CH photo-dissociation rate and Fig. 2 shows that this is indeed the case. We find an H2 formation rate constant of 2.0 × 10-17 cm3 s-1 for a-C:H/a-C grains and 4.0 × 10-17 cm3 s-1 if the a-C coated amorphous silicate grains, a-Sil/a-C, are also included. The error bar on the square data points in Fig. 2 conservatively reflects a factor of three uncertainty in the dust optical properties and a factor of ten uncertainty in ϵ.
For the small a-C grains the bulk material band gap of 0.1 eV (≡XH ~ 0.02) required by the Jones et al. (2013) model implies that these particles cannot be completely aromatised to a graphite-like material with a band gap close to 0 eV. Small a-C grains are therefore able to preserve a residual hydrogen fraction of a few percent that is seemingly hard to remove in the ISM without destroying and/or fragmenting the structure. They are therefore more resistant to UV photo-processing than expected (Jones 2012b), consistent with the H2 formation mechanism proposed in Sect. 2.
In addition to CH bond photo-dissociation by UV photons it is also possible that H2 formation could be driven by thermal desorption during small grain, whole-particle stochastic heating events. However, as discussed by (Jones 2012b, Sect. 5.1) the effects of a-C(:H) dust thermal processing may not be important. Here we do not consider thermal desorption-driven H2 formation and so our predicted formation rates may only be lower limits. Given that the small a-C grain population is sensitive to photo-processing (e.g., Pilleri et al. 2012; Jones et al. 2013), the more resistant large a-C(:H) and a-SilFe/a-C grains ought to provide a “background” H2 formation rate.
In their study Habart et al. (2004) found that in moderately excited PDRs (G0 = 102−103) the H2 formation rate is about five times the standard value but that in higher radiation field PDRs the standard value of 3 × 10-17 cm3 s-1 is sufficient. Based on their observations they also inferred a link between H2 formation and the aromatic emission band carriers. In a later study Habart et al. (2011) found that the observed column densities of rotationally excited H2 in PDRs (G0 = 5−102) are higher than that predicted by PDR models. This discrepancy could be explained by an enhancement in the H2 formation rate in PDRs (Habart et al. 2011). The observational evidence therefore seems to point towards PDRs as particularly efficient H2-forming regions when the local radiation field is moderate, which appears to be in qualitative agreement with the model proposed here.
The Jones et al. (2013) dust model predicts an enhanced abundance of small a-C grains in PDRs due to the photo-fragmentation of larger a-C(:H) grains, which is consistent with the findings of Compiègne et al. (2008) and Arab et al. (2012). This implies an enhanced H2 formation rate in moderate PDRs where the radiation field is not intense enough destroy a-C nano-particles. In very intense radiation fields (G0> 103) the destruction of the emission band carriers is observed (e.g., Boulanger et al. 1994). Here we assume that the a-C(:H) nano-particles responsible for the IR emission bands are also the source of H2 formation, and that both processes are driven by UV photon absorption. Interestingly, the work of Pilleri et al. (2012) shows that the photo-fragmentation of very small carbonaceous grains, which are PAH-based in their model, occurs in regions where H2 rotational line emission is observed. Nevertheless, extremely intense radiation fields that lead to the photo-fragmentation and destruction of a-C(:H) nano-particles (Jones et al. 2013) will close the door on H2 formation and also the IR band emission in such regions. Hence, the absolute value of G0 is a key parameter in the proposed H2 formation mechanism, which is likely to be efficient over a wide range of the G0/nH parameter space, e.g., 0.01 <G0/nH< 1.0 unless the local ISRF is extremely intense, i.e., G0 ≫ 103 (Boulanger et al. 1994).
3.3. Daughter-molecule formation
The photolysis and associated aromatisation of a-C:H materials yields decomposition products other than molecular hydrogen (Sect. 3). However, the other released species contain at least one carbon atom and result in decomposition and a structural re-adjustment to a lower hydrogen and carbon content. Smith (1984) and Alata et al. (2014) showed that a-C(:H) loses its hydrogen as H2 but also as small hydrocarbon molecules. Thus, a-C(:H) photolytic decomposition leads to significant mass loss from the material during thermal- or photo-processing. It is likely that the release of daughter radical or molecular species will be more pronounced in smaller particles (e.g., Duley 1996, 2000; Scott et al. 1997; Jones 2009). Pety et al. (2005) proposed that the small hydrocarbon molecules that they observed in PDR regions (i.e., CCH, c-C3H2, C4H)10 derive from pre-exisitng PAHs, which are undergoing UV photon-induced destruction. A similar hypothesis was proposed by Scott & Duley (1996) and extended by Jones (2009), who postulated that the precursors for the “PAHs” are themselves derived from the UV photolytic de-construction of small a-C(:H) particles in the transition regions between molecular clouds and PDRs. Such a process is supported by the laboratory work of Smith (1984), Alata et al. (2014) and Duley et al. (2015), which indicate that small molecules are formed during the degradation of bulk a-C(:H) materials. As shown by Duley et al. (2015) the primary a-C:H photo-decomposition products are hydrocarbon species (molecules and radicals) with less than four carbon atoms, e.g., C3H2, C3H4 and CnH2n−1 alkyl radicals. The experimental evidence therefore favours a top-down photo-processing route to small hydrocarbon formation in PDRs (see Sect. 2). However, Duley et al. (2015) also note the release of C6H5 (phenyl) and C6H6 (benzene) in their a-C(:H) photo-decomposition experiments, indicating that the loss of single six-fold ring systems (the smallest possible aromatic domains) also needs to be included into the aliphatic/olefinic bridge-breaking and a-C(:H) re-structuring mechanism proposed in Sect. 2.
To be viable an interstellar dust model must therefore be consistent with both the PDR observations (Pety et al. 2005, 2012; Guzmán et al. 2015) and the laboratory-observed UV photolytic-formation of small aliphatic-rich species from carbonaceous materials (e.g., Smith 1984; Scott & Duley 1996; Alata et al. 2014; Duley et al. 2015). In our recent work we used the optical and physical properties of a-C(:H) materials within the framework of a new ISM core/mantle dust model, which is qualitatively-consistent with these data (Jones 2009, 2012a,b,c; Jones et al. 2013). In this model a-C(:H) nano-particles (with a number of C atoms NC ≲ a few hundred) are of mixed aliphatic/aromatic (arophatic) composition and exhibit “cage-like” cluster structures (see Fig. 1). Such structures have been proposed as fullerene precursors (Micelotta et al. 2012) and are similar to the locally aromatic polycyclic hydrocarbons (LAPHs) proposed as IR emission band carriers by Petrie et al. (2003). The photon-driven fragmentation of such nano-particles in PDRs will disaggregate them into their their sub-component aliphatic/olefinic bridging structures (with NC ≲ 4) and small aromatics (with few rings, i.e., NR = 3−8, Jones 2012c). All of these daughter species will be more rapidly photo-dissociated in PDRs than the parent nano-particle. They are therefore transient species signalling the demise of a-C(:H) nano-particles in intense radiation fields (Jones et al. 2013) but at the same time they are the drivers of some interesting top-down interstellar chemistry.
In PDRs it is also possible that the a-C(:H) grains are undergoing a sort of slow combustion by reaction with the abundant gas phase atomic oxygen, i.e., ![Mathematical equation: \begin{equation} {\rm O_{(g)}} \ + \ {\rm [a-C(:H)]C_{(s)}} {\rm } \ \ \rightarrow \ \ {\rm [a-C(:H)]} \ + \ {\rm CO_{(g)}^\uparrow}. \label{eq_aCH_combustion} \end{equation}](/articles/aa/full_html/2015/09/aa26487-15/aa26487-15-eq127.png) (9)Such a process will lead to the formation of excited CO molecules in regions where the a-C(:H) dust is being eroded.
(9)Such a process will lead to the formation of excited CO molecules in regions where the a-C(:H) dust is being eroded.
4. Conclusions
We have shown that the UV-UV photo-processing of the aliphatic/olefinic bridging structures in arophatic a-C(:H) grains could provide a viable route to H2 formation in the ISM, where the ISRF is sufficiently intense, i.e.,  .
.
A photon-driven mechanism for H2 and daughter hydrocarbon radical and molecule formation is proposed, which operates through the chemisorption of H atoms at activated sites and CC bond destruction, respectively. It is also possible that this H2 formation mechanism could be aided by the dangling bonds favoured by low N atom doping concentrations within a-C(:H) nano-particles. The proposed process is not directly dependent on the grain temperature and therefore would provide a plausible route to H2 formation in PDRs where the large grains are warm (i.e., Tdust> 25 K) and where experiments indicate that H2 formation, involving H atom physisorption on grain surfaces, is not efficient. We note that even in the diffuse ISM (G0 = 1) the nano-particles responsible for the bulk of the H2 formation, from UV photon-induced CH bond dissociation, and also for the IR emission features have temperatures of the order of 50−70 K. In more intense radiation fields these same nano-grains will have temperatures well in excess of 100 K. In PDRs where the large grains are much warmer (Tdust ≳ 50 K), such as the Orion region, the proposed mechanism will no longer operate because the small a-C grains that dominate H2 formation are destroyed.
In high extinction regions, the UV flux will be insufficient to drive H2 formation and in extremely intense radiation fields H2 formation will be severely limited because the active sites are destroyed as the grains undergo UV photon-induced fragmentation. It therefore appears that, for a given region of the ISM, there will be an optimum set of physical conditions that will favour H2 formation by the proposed mechanism. The most favoured conditions would appear to be moderate densities and radiation fields but these will need to quantified by detailed modelling. Seemingly, PDR regions are the ideal sites for H2 formation from the photolytic processing of a-C(:H) grains.
The physical depth into a-C(:H) materials at which the optical depth is unity for VUV and EUV photons (E = 7−13.6 eV) varies little with wavelength and material composition (e.g., Jones 2012b) and we can therefore consider the collective effects of VUV and EUV photons.
The physisorption interaction energy is ~0.01–0.1 eV.
The aliphatic C−H bond energy is ~4.2–4.6 eV, while that for olefinic and aromatic C−H bonds is ~4.8–4.9 eV.
Reaction with an H atom in an aliphatic C−H bond will be favoured because of the lower bond energy, compared to olefinic and aromatic C−H bonds (see footnote 2).
As noted above we assume that a single UV photon results in the formation of an H2 molecule.
An increase in the dark, graphite-like or aromatic content in response to UV photon irradiation. Hence, the term photo-darkening is often used for the aromatisation of a-C(:H) materials.
This is the path length corresponding to an optical depth of unity for UV photons in an homogeneous, bulk a-C(:H) material. For E> 10 eV the optical depth is only weakly-dependent on the a-C(:H) material band gap and the photon energy (see Paper II). It could also perhaps be regarded as the depth into the grains at which UV photolysis is in equilibrium incident H atom re-hydrogenation.
With this definition of ϵ the experimentally-derived values of 0.014 and 0.025 (Maté 2014; Alata et al. 2014) are equivalent to a ≃ 140 and 80 nm, respectively, which can be considered as bulk materials in terms of their optical properties (Jones 2012c).
A dangling bond is an unsatisfied valence on an atom in a chemically-bonded network, which is an active chemisorption site.
To this list can now be added the ion C3H+, which has also been observed in the Horsehead Nebula (Pety et al. 2012).
Acknowledgments
The authors would like to thank Emmanuel Dartois, Nathalie Ysard and Laurent Verstraete for their thoughtful and helpful remarks on the manuscript.
References
- Adel, M. E., Amir, O., Kalish, R., & Feldman, L. C. 1989, J. Appl. Phys., 66, 3248 [NASA ADS] [CrossRef] [Google Scholar]
- Alata, I., Cruz-Diaz, G. A., Muñoz Caro, G. M., & Dartois, E. 2014, A&A, 569, A119 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Amir, O., & Kalish, R. 1991, J. Appl. Phys., 70, 4958 [NASA ADS] [CrossRef] [Google Scholar]
- Arab, H., Abergel, A., Habart, E., et al. 2012, A&A, 541, A19 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bocchio, M., Micelotta, E. R., Gautier, A.-L., & Jones, A. P. 2012, A&A, 545, A124 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Boulanger, F., Prevot, M. L., & Gry, C. 1994, A&A, 284, 956 [NASA ADS] [Google Scholar]
- Bron, E., Le Bourlot, J., & Le Petit, F. 2014, A&A, 569, A100 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Clark, B. C., Mason, L. W., & Kissel, J. 1987, A&A, 187, 779 [NASA ADS] [Google Scholar]
- Compiègne, M., Abergel, A., Verstraete, L., & Habart, E. 2008, A&A, 491, 797 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Dartois, E., Muñoz Caro, G. M., Deboffle, D., Montagnac, G., & D’Hendecourt, L. 2005, A&A, 432, 895 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Duley, W. W. 1996, MNRAS, 283, 343 [NASA ADS] [Google Scholar]
- Duley, W. W. 2000, ApJ, 528, 841 [NASA ADS] [CrossRef] [Google Scholar]
- Duley, W. W., Zaidi, A., Wesolowski, M. J., & Kuzmin, S. 2015, MNRAS, 447, 1242 [NASA ADS] [CrossRef] [Google Scholar]
- Ferrari, A. C., & Robertson, J. 2004, Phil. Trans. R. Soc. Lond. A, 362, 2477 [NASA ADS] [CrossRef] [Google Scholar]
- Godard, M., Féraud, G., Chabot, M., et al. 2011, A&A, 529, A146 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Gruzdkov, Y. A., Watanabe, K., Sawabe, K., & Matsumoto, Y. 1994, Chem. Phys. Lett., 227, 243 [NASA ADS] [CrossRef] [Google Scholar]
- Gry, C., Boulanger, F., Nehmé, C., et al. 2002, A&A, 391, 675 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Guzmán, V. V., Pety, J., Goicoechea, J. R., et al. 2015, ApJ, 800, L33 [NASA ADS] [CrossRef] [Google Scholar]
- Habart, E., Boulanger, F., Verstraete, L., Walmsley, C. M., & Pineau des Forêts, G. 2004, A&A, 414, 531 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Habart, E., Abergel, A., Boulanger, F., et al. 2011, A&A, 527, A122 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Henry, R. C. 2002, ApJ, 570, 697 [NASA ADS] [CrossRef] [Google Scholar]
- Hu, A., Griesing, S., Rybachuk, M., Lu, Q.-B., & Duley, W. W. 2007a, J. Appl. Phys., 102, 074311 [NASA ADS] [CrossRef] [Google Scholar]
- Hu, A., Lu, Q.-B., Duley, W. W., & Rybachuk, M. 2007b, J. Chem. Phys., 126, 154705 [NASA ADS] [CrossRef] [Google Scholar]
- Jones, A. P. 1990, MNRAS, 247, 305 [NASA ADS] [Google Scholar]
- Jones, A. P. 2009, in Cosmic Dust – Near and Far, eds. T. Henning, E. Grün, & J. Steinacker, ASP Conf. Ser., 414, 473 [Google Scholar]
- Jones, A. P. 2012a, A&A, 540, A1 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2012b, A&A, 540, A2 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2012c, A&A, 542, A98 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2012d, A&A, 545, C2 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2012e, A&A, 545, C3 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2013, A&A, 555, A39 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P. 2014, Planet. Space Sci., 100, 26 [NASA ADS] [CrossRef] [Google Scholar]
- Jones, A. P., Fanciullo, L., Köhler, M., et al. 2013, A&A, 558, A62 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jones, A. P., Ysard, N., Köhler, M., et al. 2014, RSC Faraday Discuss., 168, 313 [NASA ADS] [CrossRef] [Google Scholar]
- Köhler, M., Jones, A., & Ysard, N. 2014, A&A, 565, L9 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Kondo, T., Kaneda, H., Oyabu, S., et al. 2012, ApJ, 751, L18 [NASA ADS] [CrossRef] [Google Scholar]
- Li, A., & Draine, B. T. 2012, ApJ, 760, L35 [NASA ADS] [CrossRef] [Google Scholar]
- Marée, C. H. M., Vredenberg, A. M., & Habraken, F. H. P. M. 1996, Materials Chem. Phys., 46, 198 [Google Scholar]
- Maté, B. 2014, Royal Soc. Chem., Faraday Discussion, 168, 602 [Google Scholar]
- Mennella, V. 2008, ApJ, 682, L101 [NASA ADS] [CrossRef] [Google Scholar]
- Mennella, V. 2010, ApJ, 718, 867 [NASA ADS] [CrossRef] [Google Scholar]
- Mennella, V., Muñoz Caro, G. M., Ruiterkamp, R., et al. 2001, A&A, 367, 355 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Mennella, V., Hornekær, L., Thrower, J., & Accolla, M. 2012, ApJ, 745, L2 [NASA ADS] [CrossRef] [Google Scholar]
- Micelotta, E. R., Jones, A. P., & Tielens, A. G. G. M. 2010a, A&A, 510, A37 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Micelotta, E. R., Jones, A. P., & Tielens, A. G. G. M. 2010b, A&A, 510, A36 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Micelotta, E. R., Jones, A. P., Cami, J., et al. 2012, ApJ, 761, 35 [NASA ADS] [CrossRef] [Google Scholar]
- Muñoz Caro, G. M., Ruiterkamp, R., Schutte, W. A., Greenberg, J. M., & Mennella, V. 2001, A&A, 367, 347 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pendleton, Y. J., & Allamandola, L. J. 2002, ApJS, 138, 75 [NASA ADS] [CrossRef] [Google Scholar]
- Petrie, S., Stranger, R., & Duley, W. W. 2003, ApJ, 594, 869 [NASA ADS] [CrossRef] [Google Scholar]
- Pety, J., Teyssier, D., Fossé, D., et al. 2005, A&A, 435, 885 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pety, J., Gratier, P., Guzmán, V., et al. 2012, A&A, 548, A68 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pilleri, P., Montillaud, J., Berné, O., & Joblin, C. 2012, A&A, 542, A69 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pilleri, P., Joblin, C., Boulanger, F., & Onaka, T. 2015, A&A, 577, A16 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rauls, E., & Hornekær, L. 2008, ApJ, 679, 531 [NASA ADS] [CrossRef] [Google Scholar]
- Schwan, J., Dworschak, W., Jung, K., & Ehrhardt, H. 1994, Diamond and Related Materials, 3, 1034 [NASA ADS] [CrossRef] [Google Scholar]
- Scott, A., & Duley, W. W. 1996, ApJ, 472, L123 [NASA ADS] [CrossRef] [Google Scholar]
- Scott, A. D., Duley, W. W., & Jahani, H. R. 1997, ApJ, 490, L175 [NASA ADS] [CrossRef] [Google Scholar]
- Sloan, G. C., Bregman, J. D., Geballe, T. R., Allamandola, L. J., & Woodward, C. E. 1997, ApJ, 474, 735 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Smith, F. W. 1984, J. Appl. Phys., 55, 764 [NASA ADS] [CrossRef] [Google Scholar]
- Verstraete, L., Pech, C., Moutou, C., et al. 2001, A&A, 372, 981 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Welch, A. R., & Judge, D. L. 1972, J. Chem. Phys., 57, 286 [NASA ADS] [CrossRef] [Google Scholar]
- Yamagishi, M., Kaneda, H., Ishihara, D., et al. 2012, A&A, 541, A10 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
All Figures
|
Fig. 1 Schematic view of part of the 3D structure of an a-C(:H) grain containing ~60 C atoms and ~40 H atoms. Some of the aliphatic, olefinic and aromatic H atoms inherent within the structure are indicated by the red, blue and purple filled circles, respectively. Radical C atom or dangling bond sites sites are shown by the black dots. See text for details. |
| In the text | |
|
Fig. 2 Canonical interstellar H2 formation rate constant (grey horizontal band, Gry et al. 2002) compared to that predicted for H2 formation via CH bond UV photo-dissociation from a-C:H/a-C grains, kf[CH]/nH = 2.0 × 10-17 cm3 s-1 shown at a = 0.5 nm (squares with error bars). The green green square also includes H2 formation in the a-C mantles on the large a-SilFe grains into the calculation, yielding kf[CH]/nH = 4.0 × 10-17 cm3 s-1. The small squares show the relative contributions to the total H2 formation rate as a function of particle radius. The H atom sticking rate constant, for sH = 1, is shown for the small a-C (thin grey), large a-C:H/a-C (thick grey) and a-Sil/a-C (green) grain distributions and for gas temperatures of 50, 100 and 200 K (short-dashed, solid and long-dashed, respectively). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.