| Issue |
A&A
Volume 709, May 2026
|
|
|---|---|---|
| Article Number | A116 | |
| Number of page(s) | 12 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202659799 | |
| Published online | 08 May 2026 | |
Methanimine as a sink in the HCN and HNC solid state hydrogenation network
1
Leiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University,
PO Box 9502,
2300 RA
Leiden,
The Netherlands
2
Leiden Observatory, Leiden University,
PO Box 9513,
2300 RA
Leiden,
The Netherlands
★ Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
11
March
2026
Accepted:
3
April
2026
Abstract
Aims. We aim to provide a systematic and quantitative description of the hydrogenation network connecting HCN and HNC to methylamine on interstellar water ices, and to identify the dominant reaction pathways and potential bottlenecks.
Methods. We performed a comprehensive quantum-chemical investigation of H-addition, H-abstraction, reactions with H2, and water-assisted H-transfer isomerization involving all intermediates connecting HCN and HNC up to CH3NH2, considering amorphous solid water molecular clusters composed of 14 water molecules. We employed benchmarked density functional theory to derive activation barriers, elucidate the reaction mechanisms, and determine the binding energy distribution of H2CN and CNH2. We also discuss the effect of deuterium substitutions on selected points of the network.
Results. H-addition reactions are generally mediated by activation energy barriers, except for radical reactants. Considering both barriers and the associated tunneling crossover temperatures, the most favorable hydrogenation sequence starts from HNC rather than from HCN. The network initially evolves toward either methanimine (H2CNH; which emerges as the central species) or the singlet open-shell carbene 1[HCNH2]. Subsequent hydrogenation steps lead to methylamine (CH3NH2). Along these reaction paths, several processes are barrier-less (e.g., the hydrogenation of molecular radicals), while some H-abstraction reactions can compete with addition channels. Reactions involving H2 as a reactant are rare, as most of these channels are endoergic. Deuterium substitution has only a minor impact on classical activation energies but significantly affects the transition-state imaginary frequencies and, therefore, the expected tunneling efficiencies.
Conclusions. Our results support the idea that methanimine and methylamine can efficiently form from HNC on cold interstellar ices, with methanimine acting as a chemical sink within the network. HCN, on the other hand, is less reactive and hence has a higher chance of being preserved. The reaction network presented here provides quantitative constraints for astrochemical models of nitrogen-bearing organic chemistry in star-forming regions.
Key words: astrochemistry / molecular processes / ISM: molecules
© The Authors 2026
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
Numerous CN-bearing species have been detected in the interstellar medium (ISM), ranging from simple molecules such as HCN and CH3CN to larger ones such as cyanopolyynes. Owing to their large dipole moments, these molecules are readily detectable in the gas phase using radioastronomical facilities, and as a result, they are routinely employed by astronomers to trace the physical conditions of the ISM, both in the Milky Way and in external galaxies (e.g., Schilke et al. 1992; Hacar et al. 2020; Le Gal et al. 2014; Öberg et al. 2015; Jin et al. 2015; Ilee et al. 2021; Tennis et al. 2023).
From a chemical perspective, nitriles can appear as two different isomers, such as HCN and HNC or CH3CN and CH3NC. They are particularly interesting in the context of prebiotic chemistry. For example, they can hydrolyse into carboxylic acids under suitable conditions (e.g., Miller 1953; Miller & Urey 1959), a process that potentially links them to the formation of amino acids (as exemplified by the Strecker synthesis; Strecker 1854), among other prebiotic species important for the RNA-world hypothesis (see, e.g., Oró & Kamat 1961; Oró 1961; Powner et al. 2009; Patel et al. 2015; Becker et al. 2019; Cappelletti & Rahm 2026; Jiménez-Serra et al. 2020).
The simplest closed-shell CN-bearing species in the ISM are HCN and HNC, which have been extensively studied in the literature. Their relative abundances are often used as a diagnostic tool for temperature (Hacar et al. 2020), and their emission can be used to trace dense gas regions; both are critical parameters for probing the ISM and other galaxies.
Both HCN and HNC can be formed through gas-phase processes or on the surfaces of interstellar icy grains (see, e.g., Enrique-Romero & Lamberts 2024). Therefore, subsequent chemical evolution on interstellar ices should lead to more complex species. Observational evidence points to HCN surface hydrogenation as the main path to methylamine formation (e.g., Suzuki et al. 2023); this is supported by the experiments of Theulé et al. (2011), which showed that HCN hydrogenation leads to CH3NH2, with CH2NH as a likely intermediate. With computational chemistry, Woon (2002) investigated the hydrogenation of HCN and CH2NH in the context of interstellar amorphous solid water ices, using an implicit solvation model to account for the effect of the ice environment (i.e., without explicitly including water molecules). Their results show that hydrogenation at the carbon atom of HCN is significantly more favorable, with a lower activation barrier (~30.5kJ mol−11) compared to hydrogenation at the nitrogen atom (~53.5 kJ mol−1). A similar trend was observed for CH2NH, with barriers of approximately 19.2 kJ mol−1 (C-hydrogenation) and 25.5 kJ mol−1 (N-hydrogenation). More recently, Molpeceres et al. (2024) investigated the reaction of 3C + NH3 on interstellar ice surfaces. As part of their reaction network, hydrogenation pathways of HCN and HNC were also considered, although limited to atomic hydrogen and excluding molecular hydrogen (H2) reactions.
More broadly, numerous experimental studies have investigated the energetic processing of interstellar ice analogs containing cyanide-bearing molecules. Small nitriles such as HCN and CH3CN are highly reactive when exposed to UV irradiation (e.g., Hudson & Moore 2000; Gerakines et al. 2004; Bernstein et al. 2002; Danger et al. 2011b; Jiménez-Escobar et al. 2014; Bulak et al. 2021) or X-rays (e.g., Volosatova et al. 2021; Basalgète et al. 2021; Carvalho et al. 2022), as well as under ion and electron bombardment (e.g., Hudson & Moore 2000; Gerakines et al. 2004; Hudson et al. 2008). These energetic processes lead to the formation of a wide range of products, including amines, amides, aminonitriles, and salts such as OCN-, particularly when ammonia is present. A recurring observation in these experiments is that the presence of water enhances chemical complexity, while both UV and X-ray irradiation can induce desorption processes. In addition, HCN is known to undergo polymerization under energetic processing (e.g., Gerakines et al. 2004; Volosatova et al. 2021).
Despite the chemical richness observed in these experiments, the interpretation of results is often complicated by the lack of specificity in the reaction pathways. Multiple processes occur simultaneously, making it difficult to isolate individual reaction channels or derive quantitative kinetic data and branching ratios. One example of a different approach is the work of Carvalho et al. (2022), who combined soft X-ray irradiation experiments on CH3CN with a chemical network model to interpret the observed chemistry. Thanks to the use of a model and concomitant input parameters, they found that methyl cyanide can largely survive energetic processing.
In this context, theoretical studies such as the present one can help disentangle individual reaction pathways and provide the energetic information required to guide the interpretation of laboratory experiments and astrochemical models. At the same time, dedicated experiments targeting specific hydrogenation pathways of HCN and HNC on water-rich ice surfaces would be highly valuable for testing the mechanisms proposed here. To address these challenges and build upon our previous studies (Enrique-Romero & Lamberts 2024; Enrique-Romero & Lamberts 2025), we revisited the surface destruction pathways of HCN and HNC via successive hydrogen addition and abstraction reactions involving H• and H2.
We aim to connect these two molecules to their fully hydrogenated counterpart, methylamine (CH3NH2). This is in qualitative agreement with the reaction sequence proposed by the only laboratory work available in the literature, Theulé et al. (2011), who connected HCN and HNC to methylamine through methanimine. Notice that the methanimine is also relevant in the prebiotic formation of glycine (NH2CH2COOH) via the Strecker synthesis, where methanimine reacts with HCN (e.g., Danger et al. 2011a).
2 Methods
All calculations were performed using ORCA 6.0.1 (Neese et al. 2020; Neese 2022). Benchmarked density functional calculations were used across this work. Given the number of reactions studied in this work, we chose the triple-ζ-quality ma-def2-TZVP basis set (Zheng et al. 2011), balancing computational cost and accuracy for density functional calculations. We used multiple computational algorithms for this work, encompass geometry optimizations, transition state searches, intrinsic reaction coordinate calculations to connect stationary points along the reaction pathway on the potential energy surface, potential energy surface scans, nudged elastic band calculations (Ásgeirsson et al. 2021), and frequency calculations to ensure that our geometries correspond to minima or first-order saddle points for transition states. Tight self-consistent field (TightSCF) convergence criteria and dense integration grids (DEFGRID3). Unrestricted Kohn-Sham orbitals were used consistently, and for all open-shell singlet calculations, we employed the broken-spin-symmetry approach via ORCA’s spin-flip routine. A comprehensive benchmark comprising 13 reactions was performed (raw data available on Zenodo Enrique-Romero & Lamberts 2026), where the performance of a wide range of functionals in combination with the ma-def2-TZVP basis set was tested against CCSD(T)/aug-cc-pVTZ reference data. Figure 1 summarizes the results by reporting average energy deviations for selected functionals. Some functionals exhibit a marked difference in performance between H/H2 addition reactions and water-assisted H-transfer (wHt) reactions. To make this distinction clear, we report the results for these two subsets separately in Fig. 1, in addition to providing the overall average for reference. For the wHt reactions, ωB97m-D3(BJ) yields the highest accuracy. In contrast, for H/H2 additions and abstraction reactions, M06-2X-D3(0) performs best. We therefore adopted a combined strategy, applying each functional to the reaction class for which it is most reliable. In addition, the use of M06-2X-D3(0) is further supported by its successful application in our previous studies, thereby ensuring methodological consistency. From the reactions listed in Table A.1, we took H-addition reactions R1, R2, R3, R4, and R11; the H-abstraction reactions R26, R32 and R33; H2-hydrogenation reactions R22 from cis- and from trans-HCNH; and the wHt reactions R38, R39, and R40.
Finally, M06-2X-D3(0) performs poorly for the H-abstraction reactions R25 and R31, yielding unrealistic ZPE corrections, exceeding 50 kJ mol−1. For these cases, we instead employed the next-best functional from the benchmark, ωB97m-D3(BJ), which produces physically reasonable ZPE corrections, with corrected activation barriers of 8.8 and 6.6 kJ mol−1 for reactions R25 and R31. In agreement with the literature (Molpeceres et al. 2024).
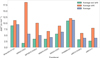 |
Fig. 1 Selected functionals from the benchmark study. Three metrics are reported: (i) the average energy deviation excluding proton-transfer reactions, (ii) the average energy deviation for proton-transfer reactions only, and (iii) the global average of all reactions. These values are obtained from 13 benchmarked reactions (see the main text for details and the appendix for tabulated data). |
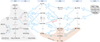 |
Fig. 2 Schematic representation of the •CN chemical network on interstellar ice surfaces. The figure summarizes the reactions investigated in this work (the hydrogenation of HCN and HNC) together with related processes reported in previous studies (Enrique-Romero et al. 2022; Molpeceres et al. 2024; Enrique-Romero & Lamberts 2024; Enrique-Romero & Lamberts 2025), shown as colored regions. Unless otherwise indicated, radical species have doublet spin multiplicity. Solid arrows denote barrier-less reactions, whereas dashed arrows indicate reactions with an activation barrier. Activation energies are given in kJ mol−1. Arrow colors indicate reaction types: blue for H addition and abstraction, red for reactions with H2, gray for wHts, and black for other processes (e.g., isomerization or radical-radical reactions). Energies are reported on a per-reaction basis, assuming efficient thermalization on amorphous solid water, such that each activation barrier is referenced to the products of the preceding step. |
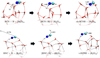 |
Fig. 3 Geometries of the H-addition reactions on HNC leading to cis- and trans-HC•NH (reactions R4-cis and R4-trans). Distances are in Å. |
3 Results
3.1 Chemical network
Four main types of reactions are considered: (i) H-additions involving atomic hydrogen, (ii) H-additions involving molecular hydrogen, (iii) H-abstractions by H atoms from molecular species (both closed-shell and radicals), and (iv) wHt isomerization reactions. To facilitate the presentation of our results, we organized the reactions by the number of H atoms added to HCN and HNC up to the five hydrogen atoms of CH3NH2, and provided the chemical network in Fig. 2. Furthermore, Table A.1, provides the complete list of reactions with complementary energetic data and a comparison with works in the literature. To aid the reader, we have also included a condensed version of the full reaction network in Fig. 8, which details the reaction pathways that we deem most important for astronomical purposes.
3.2 ne hydrogen addition
Six reactions convert HCN and HNC to species with the {H2CN} general formula. The lowest activation energy barriers correspond to HNC hydrogenation, forming either the cis- or transisomers of HC•NH (reaction R4, Fig. 3), with barriers of 8.2 and 12.5 kJ mol−1, respectively, while the barrier to form C•NH2 is 55.9 kJ mol−1. In contrast, H-addition to the more stable isomer HCN (reactions R1 and R2), involves substantially higher barriers of approximately 28.4 to form H2CN•, 33.9 kJmol−1 to form the cis-HC•NH and 41.7 for the trans-HC•NH isomer.
The HNC → HCN conversion is disfavoured, as we find water-assisted proton transfer reactions (reaction R35-wHt) exhibit large activation barriers, at least 35.1 kJ mol−1 when mediated by three water molecules, in agreement with the work by Baiano et al. (2022), where a barrier of 49.6 kJ mol−1 for the same process involving two water molecules is reported.
Given the relatively high barriers, all these reactions are expected to proceed primarily via quantum tunneling given the relatively high crossover temperatures (see Table A.1), with the lowest barrier for reaction R4-cis (HNC to cis-HC•NH). Nonetheless, these first-step hydrogenation products are likely to be short-lived intermediates, as further H-addition or abstraction reactions can take place, with either small or no barriers at all.
In summary, the more reactive isomer between HCN and HNC toward H-addition is HNC, which leads to HC•NH.
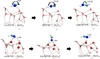 |
Fig. 4 Geometries of the isomerization reactions from cis- to trans-HC•NH (reactions R37-isom and R37-wHt). In the upper panel, the reaction coordinate is the torsion angle of HC•NH. Distances are in Å. |
3.3 Two hydrogen additions
Continuing along the hydrogenation sequence toward the {H3CN} family, the intermediates produced from the first H-addition (cis- and trans-•HCNH, C•NH2, and H2CN•), undergo further hydrogenation. Within the {H3CN} family, the most stable isomer is methanimine (H2C-NH), already hypothesized to be an intermediate along the reaction path by Theulé et al. (2011).
The H-addition reactions on {H2CN} leading to methanimine are barrier-less (reactions R8, R5). In contrast, the formation of aminocarbene and methyl nitrene involves activation barriers of 25.8 and 13.5 kJ mol−1, corresponding to reactions R9 (from the cis isomer) and R6, respectively. The remaining pathways, those involving the trans-•HCNH isomer and the reaction of C•NH2 with H• (reactions R9 and R7), are also barrier-less. The reactivity with molecular hydrogen (H2) is limited to t-HC•NH + H2 → H2CNH + H• (reaction R22), which exhibits a barrier of 36.6 kJ mol−1. Turning now toward H-abstraction reactions, the destruction of all H2CN isomers back to HCN and HNC via H-abstraction with an H atom proceeds without activation barriers.
The interconversion between cis- and trans-HCNH can proceed either via direct isomerization (torsion around the dihedral angle) or through water-assisted proton transfer (reaction R37). The direct pathway involves a barrier of 23.9 kJ mol−1, whereas the water-mediated mechanism exhibits a significantly higher barrier of 65.5 kJ mol−1 over three water molecules (see Fig. 4). In addition, two further wHt isomerization reactions were identified, namely trans-HC.NH ->[wHt][] H2CN. and C.NH2 ->[wHt][] trans-HC.NH, with activation barriers of 83.7 and 30.8 kJ mol−1, respectively.
Finally, it must be noted that methyl nitrene (3[H3CN:]) and aminocarbene (1[HC:NH2]) are higher-energy isomers of methanimine. These species exhibit non-octet electronic configurations at the nitrogen and carbon atoms, respectively, resulting in triplet and singlet ground states. To avoid ambiguity, we have explicitly indicated both their electronic state and the localization of the non-bonding electrons. Such spin configurations are characteristic of nitrenes and carbenes, where the distribution of unpaired electrons governs their reactivity and electronic structure. In the case of aminocarbene, the singlet ground state is stabilized by interaction between the lone pair of the amino group and the empty p orbital of the carbene center, which lowers its energy relative to the corresponding triplet state.
In summary, the most probable process at this stage of hydrogenation is H-addition to trans HC NH, leading preferentially to methanimine or aminocarbene formation. In contrast, the production of methyl nitrene is less likely, as it requires the hydrogenation of H2CN*, which itself forms via a barrier-mediated hydrogenation of HCN.
3.4 Three hydrogen additions
From the {H3CN} family, the main reactants involved in this third H-addition step are methanimine and aminocarbene. The hydrogenation of the former into H3CN•H or H2C•NH2 (reactions R12 and R11) exhibits activation barriers of 16.0 and 22.9 kJ mol−1, respectively. In contrast, the conversion of aminocarbene to H2C•NH2 (reaction R13, Fig. 5) proceeds with a very low barrier of only 4.0 kJ mol−1, making this the most favorable pathway. On the other hand, hydrogenation of the less relevant 3[H3CN:] isomer is barrier-less, leading directly to H3CN•H (reaction (R10)).
In addition, the isomerization through wHt from aminocarbene to methanimine sports a very low barrier, of just 4.9 kJ mol−1 over three water molecules, indicating that methanimine might be a chemical “sink” in the sense that it is a main hub in the chemical network. Finally, we found HC• NH3 to be highly unstable, as it barrier-lessy evolves into H2C•NH2 through a wHt reaction or can come back to 3[:CNH3].
Hydrogen abstraction reactions from the {H3CN} family are generally characterized by the presence of activation barriers. Methanimine is relatively resistant to destruction via this mechanism, as the only exothermic pathway leads to cis-HC•NH over a barrier of 24.8 kJ mol−1 (reaction R22). Aminocarbene can be converted back to C•NH2 (reaction R25), over a barrier of 8.8 kJ mol−1 or to trans-HC•NH (reaction R24) with a barrier of 14.8 kJ mol−1. On the other hand, methyl nitrene is more prone to being destroyed into H2CN• with a significantly lower barrier of only 6.7 kJ mol−1 (reaction R21).
In summary, forming either methanimine or aminocarbene ensures the continuation of the hydrogenation sequence rather than cycling back to the {H2CN} family. In contrast, the route involving 3[H3CN:] is of limited relevance, as its formation is kinetically unlikely and it is readily destroyed once formed.
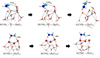 |
Fig. 5 Molecular geometries of the lowest barrier channels involving the evolution of 1[HC:NH2] into H2C•NH2 (upper panels) or its water-assisted isomerization into H2CNH (lower panels; reactions R13 and R39). Distances are in Å. |
3.5 Four hydrogen additions
The key radical intermediates H3CN•H and H2C•NH2 both undergo barrier-less reactions with atomic hydrogen to produce the fully hydrogenated product, methylamine (CH3NH2). These radicals are accessible after the hydrogenation of methylamine and aminocarbene; the latter is the faster process, given its low energy barrier. The H-abstraction reaction leading H2C•NH2 back to methanimine (reaction R29) is barrier-less, while the destruction of H3CN•H back to either methanimine or 3[H3CN:] are characterized by barriers of 4.8 and 15.6 kJ mol−1. We tried to also simulate the destruction of H2CNH2 into 1[HC:NH2], all our attempts ended up in the H-addition product, methylamine. These results further support methanimine’s role as a sink in the network. The isomerization of H3CN•H into H2C•NH2 (reaction R40) requires a high activation energy barrier of 69.3 kJ mol−1 over two water molecules.
Finally, once methylamine is formed, its destruction via H-abstraction to H3CN•H or H2C•NH2 (reactions R33 and R32) exhibit high barriers of 48.0 and 30.2 kJ mol−1, respectively. For the latter case, the nature of this barrier is related to the strong binding energy of the -NH2 group to the water ice, since, if the H atom is found right under this group, reaction R32 has a much lower activation energy barrier of just 9.9 kJ mol−1.
These results suggest that, for this final round of H-additions and abstractions, the network is strongly biased toward the accumulation of methylamine, as reverse pathways are generally hindered by activation barriers and local ice effects.
3.6 Deuteration
Deuterium substitution was investigated for a selected set of kinetically relevant reactions involving HNC and 1[HC:NH2], i.e., the energetically most favorable paths that contribute to the evolution toward methanimine and methylamine. Figure 6 summarizes the possible products arising from D-substitution for H-addition reactions. A clear kinetic isotope effect is observed only when a D atom is added, which manifests primarily in the imaginary frequencies of the transition states, with shifts on the order of about -100 cm−1, while the corresponding changes in activation energies are generally small. An exception is the reaction HNC + H → t-HC•NH, which already presents a relatively high activation barrier compared to the formation of the cis isomer and is, therefore, inherently less efficient.
A closer inspection of Fig. 6, taking into account both the energetics and the changes in barrier widths, suggests that the direct addition of a D atom to either HNC or 1[HC:NH2] is less efficient than the addition of an H atom to a parent molecule that already contains a D• substitution, under the assumption that tunneling is mandatory. From the color-coded single D-for-H substitutions, it appears that the reaction DNC + H• preferentially leads to H2CND, whereas HDCNH would more likely form via c-HC•NH + D•. Given the relatively high barrier associated with the formation of the trans-HC•NH isomer, the production of 1[HC:NH2] is more likely to proceed through the reaction 3C + NH3 Molpeceres et al. (2024). In this case, and following our previous discussion of H-addition and abstraction reactions involving H2C• NH2, it is energetically more favorable to add an H atom to the carbon center rather than abstract one of its hydrogen atoms, while abstraction from the nitrogen moiety remains feasible. Consequently, any D-substitution pattern in methylamine can be produced, whereas for methanimine, assuming the deuterium originates from NH2D, the formation of H2CND would be favored. Alternatively, if the D-addition proceeds via H2C•NH2 + D, the preferential product would be HDCNH.
Among the isomerization channels, only the wHt reaction 1[HC:NH2] → H2CNH presents a low activation barrier (4.9 kJ mol−1). As shown in Fig. 7, D substitution on 1[HC:NH2] has a negligible impact on both the activation energy and the transition-state imaginary frequency. However, D substitution on the water molecules participating in the wHt mechanism increases the barrier by approximately 2-3 kJ mol−1 and reduces the imaginary frequency by about 100-130 cm−1, indicating a higher and wider barrier.
For the wHt isomerization of HNC into HCN, a similarly weak dependence on D-substitution is observed when deuterium is initially located on DNC, although the absolute activation barriers remain comparatively high.
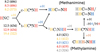 |
Fig. 6 Key D-substitution patterns for the energetically favored pathways leading to methanimine and methylamine from HNC and 1[HC:NH2]. Single deuterium substitutions are shown as color-coded H atoms. Bare values correspond to ZPE-corrected activation energies (in kJ mol−1 ; when present), and values in parentheses indicate the transition-state imaginary frequencies (|v‡|; in cm−1). Reaction steps without any activation energy value are barrier-less. |
 |
Fig. 7 Key D-substitution patterns for the wHt-mediated isomerization of HNC to HCN and 1[HC:NH2] to H2CNH over three water molecules. Single deuterium substitutions are indicated by color-coded H atoms. Bare values give ZPE-corrected activation energies (in kJ mol−1 ; when present), while values in parentheses denote the transition-state imaginary frequencies (|ν‡|; in cm−1). |
4 Discussion
4.1 Comparing to other works in the literature
Other works in the literature have partially explored the chemical network presented above, and here we provide a comparison between our results and previous ones, summarized in Table A.1. Notably, Molpeceres et al. (2024) examined several of the reaction paths presented here while investigating the condensation of carbon atoms on ammonia molecules on interstellar ices, focusing mainly on H-addition and H-abstraction reactions together with a limited number of wHt processes. While most reactions exhibit similar energetic trends in the two studies, some differences arise. These discrepancies can largely be attributed to three factors: (i) the local binding configuration on the ice surface, (ii) the influence of zero-point energy (ZPE) corrections, and (iii) methodological differences between the computational approaches.
Surface reactions on amorphous solid water depend strongly on the local binding environment, including variations in binding energies and in the hydrogen-bond network. On the one hand, for reaction R32, CH3NH2 + H• → H2C•N•H2 + H2, we identified two barriers depending on the position of the incoming H atom relative to the -NH2 group: 30.2 or 9.9 kJ mol−1 (see Fig. C.1). The higher value agrees well with literature values of 37.3 (Molpeceres et al. 2024) and 3 9.7 kJ mol−1 (Joshi & Lee 2022). The lower barrier arises from a configuration where the H atom reduces the interaction of the - NH2 moiety with the ice surface, destabilizing the reactant complex and lowering the apparent barrier, also evident from the reaction energies (see Table A.1). While this configuration is likely unstable and transient in a dynamic system (the H atom could diffuse away), it highlights the strong influence of the local ice environment on the resulting energetics. On the other hand, the wHt isomerization between 1[HC:NH2] and methanimine (reaction R39) illustrates the role of ZPE corrections. In our work, the activation barrier is only 4.9 kJ mol−1, whereas Molpeceres et al. (2024) and Ferrero et al. (2024) reported values of 22.7 and 14.0 kJ mol−1, respectively. In our calculations, the uncorrected ωB97m-D3(BJ) barrier is ~17 kJ mol−1, indicating that the ZPE correction plays a substantial role, while differences in the local hydrogen-bond environment may further contribute.
A similar case is reaction R38, for which Molpeceres et al. (2024) reported a barrier of 88.8 kJ mol−1 for formation of the trans isomer, whereas we obtain a barrier of 30.8 kJ mol−1, much closer to their gas-phase value of 39.5 kJ mol−1. The high barrier reported in that study led the authors to focus their discussion on cis-HC•NH, excluding the trans isomer. Although our value is significantly lower, this reaction is still unlikely to occur under pre-stellar conditions due to its very low tunneling crossover temperature (52 K), supporting their overall conclusions.
Molpeceres et al. (2024) were also unable to identify suitable wHt channels for the conversion of cis-HC•NH into C•NH2 and •CH2N. In their work, this limitation was attributed to the binding modes obtained for the reactants. From our perspective, however, it is more likely related to the greater geometric accessibility for H-transfer from the trans isomer compared with the cis configuration, which allows the wHt mechanism to operate more effectively.
A further discrepancy arises for reaction R29, which Molpeceres et al. (2024) considered non-viable on their ice model. In contrast, we find that this reaction proceeds without an activation barrier, with a reaction energy of 273.5 kJ mol−1, consistent with the gas-phase calculations of Joshi & Lee (2022). This difference likely reflects the difficulty of locating transition states for this type of reaction on the ice surface.
Finally, while HCN is less reactive than HNC against H-addition reactions in our network, one could ask whether reactions with other radicals might lead to a different conclusion. Indeed, although direct HCN + radical chemistry on interstellar ices remains largely unexplored, the study of Boland et al. (2024) still reinforces this selectivity: OH-addition pathways differ strongly for HCN and HNC and generally proceed more readily from HNC.
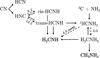 |
Fig. 8 Simplified reaction network showing the main steps along the most efficient path in the formation of methanimine and methylamine. Unless specified, the arrows indicate hydrogenation or hydrogen abstraction steps. Solid lines indicate barrier-less channels, while dashed ones indicate the presence of an activation barrier. The entrance channel through 3C + NH3 involves a minimum of two steps; see the main network in Fig. 2. |
4.2 Astrophysical implications
Our results on the hydrogenation of HCN and HNC on interstellar ices reveal a network comprising both barrier-less and barrier-mediated steps, many of which are likely enhanced by quantum tunneling. A key outcome is that HCN is expected to survive longer than HNC under interstellar conditions, owing to the higher reactivity of HNC toward atomic hydrogen. For clarity, a simplified representation of the chemical network is provided in Fig. 8, which serves as a guide throughout the following discussion.
As summarized in Fig. 8, hydrogenation preferentially proceeds from HNC → HC•NH. From there, the system can evolve toward 1[HC:NH2] → H2C•NH2 → CH3NH2 or remain at the methanimine stage since the alternative route H2CNH → H3CN•H → CH3NH2 is energetically less favorable. Starting from HNC, formation of HC•NH is associated with activation barriers of ~8 and 13 kJ mol−1, depending on the conformation of the product. In addition, H-abstraction reactions can also occur, and in cases that involve a radical, they are barrierless. This indicates that the relative orientation of the encounter between radicals and H atoms plays an important role in determining the reaction outcome, and suggests the presence of H-addition/H-abstraction loops within the network. Methanimine may act as a chemical sink, in the sense that one more step forward of backward in the network is mediated by a sizable activation energy barrier (of around 25 kJ mol−1 ), and therefore would require efficient quantum tunneling, while other intermediates such as 1[HC:NH2] show a small barrier to the next step (4.0 kJ mol−1 ). In addition, according to our calculations, wHt isomerizations between methanimine and 1[HC:NH2] are only exothermic in the direction toward the formation of methanimine, with a low activation energy (4.9 kJ mol−1).
One may ask what the importance of reactions involving molecular hydrogen (H2) is. Here we observe that not all radical + H2 → molecule + H are exoergic and therefore cannot take place under interstellar conditions. The only case where H2 participates as a reactant with a mild barrier is trans-HC•NH + H2 → H2CNH (with a barrier of 36.6 kJ mol−1), still rather high. Therefore, the presence of free H atoms in the environment is rather important to fuel the network in Fig. 2. In any case, other channels exist that can lead to the formation of methanimine and methylamine, such as radical-radical chemistry (e.g., CH3 +NH2 Garrod et al. 2008; Enrique-Romero et al. 2022), gas-phase processes (e.g., Turner et al. 1999; Sil et al. 2018; Luthra et al. 2023), or a mix of the two (such as via H-abstraction from methylamine Joshi & Lee 2022). Similarly, the further evolution of methanimine and methylamine is not limited to surface chemistry, but gas-phase chemistry can also play an important role (Vazart et al. 2015; Vazart et al. 2016; Lupi et al. 2020; de Jesus et al. 2021).
In addition to the final products of the hydrogenation network, there are intermediates that are worth exploring in more detail. San Andrés et al. (2023) propose that H2CN•/C•NH2 can be used as temperature tracers, much like HCN/HNC is routinely used (Hacar et al. 2020). Looking at our network and the one by Molpeceres et al. (2024), H2CN• is much more likely to be produced from the hydrogenation of HCN, while C•NH2 seems to be a product of an H-abstraction on 3[:CNH3], formed from 3C + NH3. The isomerization between these two isomers seems rather ineffective, since wHt reactions favor the formation of trans-HC•NH, and in addition, they can easily be destroyed by H-additions or abstractions. In any case, we calculated their binding energy distributions following the same approach as in Enrique-Romero & Lamberts (2024, see our Fig. B.1), finding that, on average, C• NH2 binds more strongly to the water ice matrix than H2CN•. The former shows a bimodal distribution with two peaks at around 42 and 58 kJ mol−1, while the latter sports a single peak at ~22 kJ mol−1. In summary, these two species, if produced on interstellar ices, are quite likely to come from different direct parent species in our network.
Methanimine and methylamine are important intermediates in astrochemistry, as both are potential precursors to pre-biotically relevant molecules such as glycine (NH2CH2COOH; Bernstein et al. 2002; Elsila et al. 2007). In any case, the H2C• NH2 radical may play an important role in glycine formation through the radical-radical coupling with the HOC•O radical, as proposed in the reaction: HO•CO + •CH2NH2 → NH2CH2COOH (Woon 2002). Methanimine is both efficiently formed and relatively stable, and it is therefore expected to accumulate on interstellar ice surfaces. As a result, methanimine may serve as a key precursor to glycine formation. Although conversion to the H2C•NH2 radical requires overcoming a moderately high activation barrier (22.9 kJ mol−1), the enhanced abundance of methanimine can still facilitate its involvement in downstream glycine-forming pathways.
Finally, we examined the effects of deuterium substitution along the main formation routes to methanimine and methylamine (see Sect. 3.6). Overall, D substitution has only a minor impact on activation energies but leads to lower imaginary frequencies, indicating slightly wider effective barriers and thus reduced tunneling efficiency for reactions involving direct D addition. As a result, H-addition pathways remain kinetically favored, although the overall structure of the network is not altered. From an astrochemical perspective, this implies that multiple deuteration pathways remain viable. In the case of methanimine, both DNC-derived routes and pathways involving NH2D can contribute, while the weak sensitivity of wHt reactions to isotopic substitution suggests that exchange with the ice matrix can efficiently redistribute deuterium. Consequently, we do not expect that the presence of singly deuterated methanimine uniquely constrains its formation pathway. A similar situation applies to methylamine. Its formation through successive hydrogenation and abstraction steps allows multiple D-substitution patterns, with their relative abundances primarily governed by the local availability of deuterated precursor species rather than by a single dominant mechanism. All of this leads to the conclusion that the most likely deuterated products are H2CND (linked to DNC), HDCNH, and CH3NHD (linked with NH2D) and CH2DNH2.
5 Conclusions
In this work we explored the hydrogenation network connecting HCN and HNC to methylamine on interstellar water ices. A schematic overview of the network is provided in Fig. 8. Our calculations show that HNC constitutes the most favorable entry point into the hydrogenation sequence, as H addition leading to cis-HC*NH proceeds over significantly lower barriers than the corresponding reactions involving HCN. Water-assisted HNC→HCN isomerization is also unlikely to compete under cold pre-stellar conditions due to the high associated barriers. These results suggest that HCN can survive longer on interstellar ice surfaces than its isomer HNC.
Within the network, methanimine (H2CNH) occupies a central position because several pathways converge toward this intermediate. However, its further hydrogenation requires overcoming moderate activation barriers, which may slow its conversion and effectively make it a chemical sink under interstellar conditions. The story is different for the two higher-energy isomers of methanimine. On the one hand, aminocarbene (1[HC:NH2]) provides an efficient route toward full hydrogenation. The conversion of this intermediate to H2C*NH2 proceeds over a very small barrier (4.0 kJ mol−1), facilitating rapid progression toward methylamine, whereas pathways leading to triplet methyl nitrene (3[H3CN:]) are disfavored.
In any case, if the radical intermediates H3CN*H or H2C*NH2 are formed, the final hydrogenation step leading to methylamine (CH3NH2) is barrier-less, making the final saturation efficient under H-rich conditions. Conversely, H-abstraction from methylamine generally involves sizable barriers. Reactions involving molecular hydrogen are instead mostly inefficient, as they are associated with comparatively high activation barriers or are endoergic, highlighting the dominant role of atomic hydrogen in driving this chemistry during the early stages of star formation.
The local ice environment strongly influences reactivity. For instance, differences in binding energies can lead to less stable reactant configurations with lower relative activation barriers compared to cases where the adsorbate resides in a deeper binding site. Similarly, for wHt reactions, the structure of the hydrogen-bond network, such as the number of water molecules involved in the transference of the H atom, plays a critical role in determining the reaction energetics. Finally, we have also investigated the effect of deuterium substitution, which mainly affects barrier widths associated with tunneling efficiencies, having only a limited impact on barrier heights. Additional supporting information and optimized molecular geometries are available in the online repository (Enrique-Romero & Lamberts 2026).
Acknowledgements
This project has received funding from the Horizon Europe Framework Programme (HORIZON) under the Marie Sklodowska-Curie grant agreement No 101149067, “ICE-CN”. Finally, this work was granted access to the HPC resources of the high-performance computer SNELLIUS, part of the SURF cooperative of educational and research institutions in the Netherlands, under project No. EINF-12426.
References
- Ásgeirsson, V., Birgisson, B. O., Bjornsson, R., et al. 2021, J. Chem. Theory Comput., 17, 4929 [CrossRef] [Google Scholar]
- Baiano, C., Lupi, J., Barone, V., & Tasinato, N. 2022, J. Chem. Theory Computat., 18, 3111 [Google Scholar]
- Basalgète, R., Ocaña, A. J., Féraud, G., et al. 2021, ApJ, 922, 213 [CrossRef] [Google Scholar]
- Becker, S., Feldmann, J., Wiedemann, S., et al. 2019, Science, 366, 76 [NASA ADS] [CrossRef] [Google Scholar]
- Bernstein, M. P., Dworkin, J. P., Sandford, S. A., Cooper, G. W., & Allamandola, L. J. 2002, Nature, 416, 401 [Google Scholar]
- Boland, M., Chaquin, P., Volatron, F., & Markovits, A. 2024, A&A, 682, A13 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bulak, M., Paardekooper, D. M., Fedoseev, G., & Linnartz, H. 2021, A&A, 647, A82 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cappelletti, M., & Rahm, M. 2026, J. Am. Chem. Soc., 148, 3949 [Google Scholar]
- Carvalho, G. A., Pilling, S., & Galvão, B. R. L. 2022, MNRAS, 515, 3760 [Google Scholar]
- Danger, G., Borget, F., Chomat, M., et al. 2011a, A&A, 535, A47 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Danger, G., Bossa, J. B., de Marcellus, P., et al. 2011b, A&A, 525, A30 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- de Jesus, D. N., da Silva, J. M. B. A., Tejero, T. N., et al. 2021, MNRAS, 501, 1202 [Google Scholar]
- Elsila, J. E., Dworkin, J. P., Bernstein, M. P., Martin, M. P., & Sandford, S. A. 2007, ApJ, 660, 911 [NASA ADS] [CrossRef] [Google Scholar]
- Enrique-Romero, J., & Lamberts, T. 2024, J. Phys. Chem. Lett., 15, 7799 [Google Scholar]
- Enrique-Romero, J., & Lamberts, T. 2025, A&A, 699, A235 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Enrique-Romero, J., & Lamberts, T. 2026, https://doi.org/10.5281/zenodo.17243174 [Google Scholar]
- Enrique-Romero, J., Rimola, A., Ceccarelli, C., et al. 2022, ApJS, 259, 39 [NASA ADS] [CrossRef] [Google Scholar]
- Ferrero, S., Ceccarelli, C., Ugliengo, P., Sodupe, M., & Rimola, A. 2024, ApJ, 960, 22 [NASA ADS] [CrossRef] [Google Scholar]
- Garrod, R. T., Widicus Weaver, S. L., & Herbst, E. 2008, ApJ, 682, 283 [Google Scholar]
- Gerakines, P. A., Moore, M. H., & Hudson, R. L. 2004, Icarus, 170, 202 [Google Scholar]
- Hacar, A., Bosman, A. D., & van Dishoeck, E. F. 2020, A&A, 635, A4 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Hudson, R. L., & Moore, M. H. 2000, A&A, 357, 787 [NASA ADS] [Google Scholar]
- Hudson, R. L., Moore, M. H., Dworkin, J. P., Martin, M. P., & Pozun, Z. D. 2008, Astrobiology, 8, 771 [Google Scholar]
- Ilee, J. D., Walsh, C., Booth, A. S., et al. 2021, ApJS, 257, 9 [NASA ADS] [CrossRef] [Google Scholar]
- Jiménez-Escobar, A., Giuliano, B. M., Muñoz Caro, G. M., Cernicharo, J., & Marcelino, N. 2014, ApJ, 788, 19 [CrossRef] [Google Scholar]
- Jiménez-Serra, I., Martin-Pintado, J., Rivilla, V. M., et al. 2020, Astrobiology, 20, 1048 [CrossRef] [Google Scholar]
- Jin, M., Lee, J.-E., & Kim, K.-T. 2015, ApJS, 219, 2 [Google Scholar]
- Joshi, P. R., & Lee, Y.-P. 2022, Commun. Chem., 5, 62 [CrossRef] [Google Scholar]
- Le Gal, R., Hily-Blant, P., Faure, A., et al. 2014, A&A, 562, A83 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Lupi, J., Puzzarini, C., & Barone, V. 2020, ApJL, 903, L35 [Google Scholar]
- Luthra, H., Wakelam, V., Sharma, M. K., & Chandra, S. 2023, MNRAS, 521, 2181 [Google Scholar]
- Majumdar, L., Loison, J. C., Ruaud, M., et al. 2018, MNRAS, 473, L59 [NASA ADS] [CrossRef] [Google Scholar]
- Miller, S. L. 1953, Science, 117, 528 [Google Scholar]
- Miller, S. L., & Urey, H. C. 1959, Science, 130, 245 [NASA ADS] [CrossRef] [Google Scholar]
- Molpeceres, G., Tsuge, M., Furuya, K., et al. 2024, J. Phys. Chem. A, 128, 3874 [NASA ADS] [CrossRef] [Google Scholar]
- Neese, F. 2022, WIREs Computat. Mol. Sci., 12 [Google Scholar]
- Neese, F., Wennmohs, F., Becker, U., & Riplinger, C. 2020, J. Chem. Phys., 152, 224108 [NASA ADS] [CrossRef] [Google Scholar]
- Öberg, K. I., Guzmán, V. V., Furuya, K., et al. 2015, Nature, 520, 198 [Google Scholar]
- Oró, J. 1961, Nature, 191, 1193 [Google Scholar]
- Oró, J., & Kamat, S. 1961, Nature, 190, 442 [Google Scholar]
- Patel, B. H., Percivalle, C., Ritson, D. J., Duffy, C. D., & Sutherland, J. D. 2015, Nat. Chem., 7, 301 [Google Scholar]
- Powner, M. W., Gerland, B., & Sutherland, J. D. 2009, Nature, 459, 239 [NASA ADS] [CrossRef] [Google Scholar]
- San Andrés, D., Colzi, L., Rivilla, V. M., et al. 2023, MNRAS, 523, 3239 [Google Scholar]
- Schilke, P., Walmsley, C. M., Pineau Des Forets, G., et al. 1992, A&A, 256, 595 [Google Scholar]
- Sil, M., Gorai, P., Das, A., et al. 2018, ApJ, 853, 139 [NASA ADS] [CrossRef] [Google Scholar]
- Strecker, A. 1854, Liebigs Ann., 91, 349 [Google Scholar]
- Suzuki, T., Majumdar, L., Goldsmith, P. F., et al. 2023, ApJ, 954, 189 [NASA ADS] [CrossRef] [Google Scholar]
- Talbi, D., & Ellinger, Y. 1996, Chem. Phys. Lett., 263, 385 [NASA ADS] [CrossRef] [Google Scholar]
- Tennis, J. D., Xue, C., Talbi, D., et al. 2023, MNRAS, 525, 2154 [Google Scholar]
- Theulé, P., Borget, F., Mispelaer, F., et al. 2011, A&A, 534, A64 [Google Scholar]
- Turner, B. E., Terzieva, R., & Herbst, E. 1999, ApJ, 518, 699 [NASA ADS] [CrossRef] [Google Scholar]
- Vazart, F., Latouche, C., Skouteris, D., Balucani, N., & Barone, V. 2015, ApJ, 810, 111 [NASA ADS] [CrossRef] [Google Scholar]
- Vazart, F., Calderini, D., Puzzarini, C., Skouteris, D., & Barone, V. 2016, J. Chem. Theo. Comp., 12, 5385 [Google Scholar]
- Volosatova, A. D., Lukianova, M. A., Zasimov, P. V., & Feldman, V. I. 2021, Phys. Chem. Chem. Phys., 23, 18449 [Google Scholar]
- Woon, D. E. 2002, ApJL, 571, L177 [Google Scholar]
- Zheng, J., Xu, X., & Truhlar, D. G. 2011, Theo. Chem. Acc., 128, 295 [Google Scholar]
Appendix A Reaction energetic descriptors
Reaction energetic descriptors and comparison to the literature.
Appendix B BE distributions of CNH2 and CH2N
The binding energy distributions for H2CN• and C•NH2 are shown in Fig. B.1. Representative examples of the most common interaction modes are presented in Fig. B.2, including one case for H2CN and two for CNH2. The first two structures correspond to adsorption on the same binding site, illustrating how small structural differences, specifically the position of the two H atoms in the admolecule, can lead to significant differences in binding energy. The third case corresponds to a deep binding site for CNH2, where the NH2 group is embedded within a small pocket-like structure in the ice, resulting in a substantially stronger interaction.
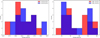 |
Fig. B.1 Binding energy distributions for H2CN (left) and CNH2 (right) on 14 and 18 water ice molecule clusters. |
Appendix C Effect of the binding mode on activation energy barriers
To illustrate the influence of the local binding configuration on the activation barrier, Fig. C.1 shows the stationary points corresponding to reaction R32 discussed in Sect. 4. Depending on the relative position of the incoming H atom with respect to the -NH2 group of methylamine, two different activation barriers are obtained. When the H atom resides directly beneath the -NH2 group, the interaction of the amine moiety with the ice surface is weakened, destabilizing the reactant complex and resulting in a lower barrier (9.9 kJ mol−1). In contrast, when the H atom originates from a different binding site on the surface and does not perturb the interaction of CH3NH2 with the ice, the reactant configuration remains more stable, and the activation barrier increases to 30.2kJmol−1.
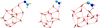 |
Fig. B.2 Geometries of the most frequent binding energies in Fig. B.1. From left to right: H2CN (22.1 kJ mol-1), CNH2 (42.5 and 55.9 kJ mol-1), and (H2O)14. |
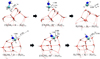 |
Fig. C.1 Stationary points along the potential energy surface for the H-abstraction reaction CH3NH2 + H• → H2C•NH2 + H2. The upper panel shows the configuration where the incoming H atom is located beneath the -NH2 group, weakening its interaction with the ice surface and leading to a lower activation barrier (9.9 kJ mol−1). The lower panel shows a configuration in which the H atom originates from a different binding site and does not perturb the interaction of CH3NH2 with the surface, resulting in a higher barrier (30.2 kJ mol−1). Bond distances are in Å. |
Appendix D Benchmark data
A selection of 13 reactions was chosen to run a benchmark study. Table D.1 contains their relative energetics (barriers and reaction energies) at the reference level. The relative differences of their activation energy with different functionals (in absolute value) can be found in D.2. Raw energies can be obtained in the Zenodo repository of this project, Enrique-Romero & Lamberts (2026).
Benchmark reference values.
Benchmark energetic descriptors.
To convert kJ mol−1 into K, one only needs to multiply by 120.27 K/kJ mol−1.
All Tables
All Figures
|
Fig. 1 Selected functionals from the benchmark study. Three metrics are reported: (i) the average energy deviation excluding proton-transfer reactions, (ii) the average energy deviation for proton-transfer reactions only, and (iii) the global average of all reactions. These values are obtained from 13 benchmarked reactions (see the main text for details and the appendix for tabulated data). |
| In the text | |
|
Fig. 2 Schematic representation of the •CN chemical network on interstellar ice surfaces. The figure summarizes the reactions investigated in this work (the hydrogenation of HCN and HNC) together with related processes reported in previous studies (Enrique-Romero et al. 2022; Molpeceres et al. 2024; Enrique-Romero & Lamberts 2024; Enrique-Romero & Lamberts 2025), shown as colored regions. Unless otherwise indicated, radical species have doublet spin multiplicity. Solid arrows denote barrier-less reactions, whereas dashed arrows indicate reactions with an activation barrier. Activation energies are given in kJ mol−1. Arrow colors indicate reaction types: blue for H addition and abstraction, red for reactions with H2, gray for wHts, and black for other processes (e.g., isomerization or radical-radical reactions). Energies are reported on a per-reaction basis, assuming efficient thermalization on amorphous solid water, such that each activation barrier is referenced to the products of the preceding step. |
| In the text | |
|
Fig. 3 Geometries of the H-addition reactions on HNC leading to cis- and trans-HC•NH (reactions R4-cis and R4-trans). Distances are in Å. |
| In the text | |
|
Fig. 4 Geometries of the isomerization reactions from cis- to trans-HC•NH (reactions R37-isom and R37-wHt). In the upper panel, the reaction coordinate is the torsion angle of HC•NH. Distances are in Å. |
| In the text | |
|
Fig. 5 Molecular geometries of the lowest barrier channels involving the evolution of 1[HC:NH2] into H2C•NH2 (upper panels) or its water-assisted isomerization into H2CNH (lower panels; reactions R13 and R39). Distances are in Å. |
| In the text | |
|
Fig. 6 Key D-substitution patterns for the energetically favored pathways leading to methanimine and methylamine from HNC and 1[HC:NH2]. Single deuterium substitutions are shown as color-coded H atoms. Bare values correspond to ZPE-corrected activation energies (in kJ mol−1 ; when present), and values in parentheses indicate the transition-state imaginary frequencies (|v‡|; in cm−1). Reaction steps without any activation energy value are barrier-less. |
| In the text | |
|
Fig. 7 Key D-substitution patterns for the wHt-mediated isomerization of HNC to HCN and 1[HC:NH2] to H2CNH over three water molecules. Single deuterium substitutions are indicated by color-coded H atoms. Bare values give ZPE-corrected activation energies (in kJ mol−1 ; when present), while values in parentheses denote the transition-state imaginary frequencies (|ν‡|; in cm−1). |
| In the text | |
|
Fig. 8 Simplified reaction network showing the main steps along the most efficient path in the formation of methanimine and methylamine. Unless specified, the arrows indicate hydrogenation or hydrogen abstraction steps. Solid lines indicate barrier-less channels, while dashed ones indicate the presence of an activation barrier. The entrance channel through 3C + NH3 involves a minimum of two steps; see the main network in Fig. 2. |
| In the text | |
|
Fig. B.1 Binding energy distributions for H2CN (left) and CNH2 (right) on 14 and 18 water ice molecule clusters. |
| In the text | |
|
Fig. B.2 Geometries of the most frequent binding energies in Fig. B.1. From left to right: H2CN (22.1 kJ mol-1), CNH2 (42.5 and 55.9 kJ mol-1), and (H2O)14. |
| In the text | |
|
Fig. C.1 Stationary points along the potential energy surface for the H-abstraction reaction CH3NH2 + H• → H2C•NH2 + H2. The upper panel shows the configuration where the incoming H atom is located beneath the -NH2 group, weakening its interaction with the ice surface and leading to a lower activation barrier (9.9 kJ mol−1). The lower panel shows a configuration in which the H atom originates from a different binding site and does not perturb the interaction of CH3NH2 with the surface, resulting in a higher barrier (30.2 kJ mol−1). Bond distances are in Å. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.