| Issue |
A&A
Volume 682, February 2024
|
|
|---|---|---|
| Article Number | A13 | |
| Number of page(s) | 5 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202348171 | |
| Published online | 29 January 2024 | |
Formamide synthesis in the interstellar medium catalyzed by damaged water ice
Sorbonne Université, Laboratoire de Chimie Théorique,
UMR 7616 CNRS,
75005
Paris,
France
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
5
October
2023
Accepted:
10
November
2023
Abstract
Context. Formamide is one of the possible precursors of life because it has a bond analogous to the peptide bond.
Aims. In this work, we examine the reaction pathways that lead from HCN or HNC and OH to formamide. Both HCN and HNC are present in the interstellar medium, while OH could be present in interstellar water ice, which under the effect of cosmic rays, partially decomposes into H and OH.
Methods. We carried out first principles calculations. We represented the solid state either by a model of clusters or by a model of slabs that takes into account periodicity. The confrontation of these two models and with the reaction in the gas phase enabled us to find reactivity trends.
Results. For HCN, the formation of the C-N bond presents an energy barrier that cannot be overcome in the interstellar medium. The presence of water ice grains does not catalyze this step. The formation of the same bond from HNC is spontaneous, even without the presence of the solid. The second step of the pathway is a transposition of H. This step requires the presence of water ice for the barrier to allow the reaction to take place in the interstellar medium. The last step is a hydrogenation of a barrier-free radical. Our work therefore concludes that the synthesis of formamide can take place in the interstellar medium through water ice, which not only catalyzes the reaction but also constitutes a reservoir of OH.
Key words: astrochemistry / methods: numerical / ISM: abundances / astrobiology
© The Authors 2024
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
For several decades, observational data from the interstellar medium have shown the presence of complex organic molecules (COMs). It is now established that molecular complexification takes place in the interstellar medium, despite the very low temperature and extreme dilution of matter. When it comes to prebiotic molecules, understanding this complexification is essential for studying the origin of life. This is a very active area of research in astrochemistry and in theoretical chemistry (Pérez-Villa et al. 2020). Among the prebiotic molecules, formamide has a prominent place (Herbst & Van Dishoeck 2009; Caselli & Ceccarelli 2012; Ceccarelli et al. 2017; Saladino et al. 2012). Indeed, formamide contains the sequence -CO-NH-, which recalls the peptide bond. Formamide could therefore be a precursor of amino acids. The same issue for other COMs also applies to formamide: There is a controversy between the proponents of a gas-phase formation (Puzzarini & Barone 2020) and those who favor catalysis on the grains of the interstitial medium (Chiavassa et al. 2005). The number of studies on the formation of formamide is enormous. The quantum calculations of Barone et al. (2015) have shown that the reaction NH2 + H2CO → NH2CHO + H is barrierless in the gas phase. More recently, other works have discussed this pathway for formamide formation (Martínez-Bachs & Rimola 2021; Douglas et al. 2022). In laboratory experiments, Dulieu et al. (2019) obtained formamide via hydrogenation of NO and H2CO• on interstellar ice analogues. Rimola et al. (2018) explored the reactivity on water ice grains through quantum calculations and water clusters. Of the three reaction paths considered, only the reaction of the CN adsorbed on a water ice surface yielded formamide. In the envisaged reaction path, water molecules are not an inert support whose only possible role is to concentrate species from the interstellar medium or lower energetic barriers. On the contrary, water molecules react to CN radicals. Moreover, water molecules catalyze the hydrogen transfer necessary to obtain the formamide. Other authors (Darla et al. 2019) have also found that it is possible to obtain formamide from the reaction HCN + H2O. In the reaction mechanism, one water molecule initially participates in the reaction, and a second acts as a catalyst. The structure of water ice in the interstellar medium has not yet been clearly identified. However, this water ice is subjected to cosmic rays. Under this effect, some water molecules are destroyed and give rise to, among other things, OH and H radicals before they recombine into H2O (Redondo et al. 2020, 2021). We therefore propose to add these radicals in water ice models. Like all radicals, OH and H are particularly reactive chemical species. We thus assumed that they can react with another species of the interstellar medium to give formamide. In such a case, HCN or HNC are excellent candidates. HCN (Snyder & Buhl 1971) and HNC (Zuckerman et al. 1972) have been detected in various objects in which the HCN/HNC ratio varies greatly (Fuente et al. 2003; Irvine & Schloerb 1984; Schilke et al. 1992; Tennekes et al. 2006).
2 Computational strategy
We used two kinds of models to represent solid water and study its influence on each reaction step. We could therefore draw conclusions from two different approaches. The first model is a cluster model. In this approach, the gas-phase reactions are compared to those where the reactants are put in the presence of one or two water molecules. At first sight, this model might seem oversimplified. Nevertheless, it is reasonable to think that a model is in itself a simplification that allows a trend to be identified. Moreover, water ice is a molecular crystal and therefore has specific characteristics. One of them, to our advantage, is that there is no band structure with a large dispersion. In this cluster model, which represents a solid, we could investigate which positions of atoms of the water ice should be optimized, and we decided to leave everything free. This model could provide a local representation of amorphous water ice.
The second model takes into account the periodicity of the solid. A cell is reproduced in all three directions of space. The norm of the vector perpendicular to the surface is large enough to make the interaction between a slab and its translational image negligible. There is at least 10 Å of void between the top of one slab with its adsorbate and the bottom of the image by translation symmetry. In this model, it is important to know the number of layers of atoms or molecules necessary to obtain the converged value of the property studied. The chosen surface is the boat surface of the apolar hexagonal ice Ih. We performed tests with different slabs. Perpendicularly to the surface studied, one can distinguish an alternation of close and distant oxygen planes. For two close oxygen planes, we use the term “bilayers”. Table 1 shows the evolution of the HCN interaction energy as a function of the number of bilayers and the number of bilayers whose geometry has been optimized. The adsorption energy is defined as the difference between the energy of the super system and the sum of the energies of the isolated adsorbate and the water ice slab. Our convention is that the adsorption energy is positive for an exothermic interaction. The results showed that there is no need to choose a thick slab with several optimized ice layers. To avoid an extra computational cost due to the hybrid functional used (see below), our slab is composed of one frozen bilayer. This choice seems reasonable given that there is already an uncertainty of the result because of the choice of the functional and the accuracy we use to draw conclusions. Nevertheless, the conclusions we draw take into account an uncertainty of several kilocalories per mol. The cell parameters, a = 8.8 Å and b = 14.6 Å, were optimized. The supercell chosen renders the interactions between adsorbates of neighboring cells negligible. Regarding the quantum methodology for the gas phase calculations, we performed calculations at the CCSD(T) level with the cc-pVTZ basis set using the geometries obtained at the DFT level with the widely used hybrid B3LYP Becke 3-parameter Lee– Yang-Parr (Becke 1992; Lee et al. 1988) functional. These allowed for consideration of the correlation and dispersion effects and are used as a reference. All cluster calculations were done using the Gaussian09 package (Caricato et al. 2009). The slab calculations were performed using the ab-initio total-energy code VASP (Kresse & Furthmiiller 1996; Kresse & Joubert 1999).
Spin-polarized approximation calculations and plane-wave basis sets with a kinetic cutoff of 400 eV describe the valence electrons. The core electrons were replaced by projector augmented wave (PAW) pseudopotentials (Blöchl 1994). The relaxation of the positions of the atoms stopped when all forces were smaller than 0.001 eV/Å. The Grimme correction (Grimme et al. 2010) to density functionals accounted for the dispersion interactions specific to this type of system. The calculations were performed by sampling the Brillouin zone in 3 × 2 × 1 Monkhorst-Pack set. As the CCSD(T) calculations were not possible, we had to choose a functional. For this, we used the addition reaction of OH on HCN in the gas phase (HO + HCN → HOHC=N) as a benchmark. The description of radical systems requires the use of hybrid functionals. The reaction energy (i.e., the energy difference between the product and the reactants) is given in Table 2. We used the Perdew-Burke-Ernzerhof (PBE; Ernzerhof & Scuseria 1999) functional with different weights for the exact Hartree-Fock exchange. The PBE is indeed a functional often used in periodic codes. We also tried the very popular B3LYP (Becke 1992; Lee et al. 1988) functional, which has a weight of 20% for the exact Hartree-Fock exchange. The result obtained with B3LYP, -26.0 kcalmol-1, is very close to the reference one (CCSD(T)/cc-pVTZ, -28,7 kcalmol-1; see Table 2). The same is true for the kinetic barrier with the zero point energy (ZPE) correction: 2.8 kcalmol-1 in B3LYP with VASP against 3.9 kcal mol CCSD(T)-pVTZ.
The VASP calculations with hybrid functionals are extremely costly. Therefore, we performed the geometry optimizations using the pure GGA PBE functional, and then, using the obtained geometry, a pointwise calculation allowed us to compute the energies given in the rest of this article. Thus, the energy variation with the ZPE correction obtained by this strategy for the reaction HO + HCN • HOHCN is –29.2 kcalmol-1, compared to –29.0 kcalmol-1 obtained with a B3LYP geometry optimization. In this work, we use the cluster model and the slab model in a complementary way. Reactivity trends were derived from the results of the cluster calculations, and the decisive steps of the reaction paths considered are identified. The most important steps are also analyzed using the periodic model.
Adsorption energy (kcalmol-1) of HCN on the boat structure surface of hexagonal water ice Ih according to the number of bilayers and the number of bilayers for which the geometry is optimized.
Energy reaction (kcalmol-1) of the HO + HCN = HOHCN reaction calculated using the VASP code and PBE hybrid functionals with variable weights of the exact Hartree-Fock exchange.
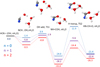 |
Fig. 1 Reaction profiles for HCN + OH → •NH-C=O (CCSD(T)/cc-pvTZ//B3LYP/cc-pVTZ + ZPE). |
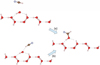 |
Fig. 2 Attack of HCN on OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice without hydrogen bonding (path a) and with (path b). |
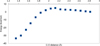 |
Fig. 3 Potential energy surface of the addition of HCN to OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice (path a). The reference is the separate fragments. |
3 Results and discussion
3.1 The case of HCN as a reactant
We assumed that the steps are as follows. The first one is the OH addition on HCN: OH• + HCN → HOCHN•. The second step is the H transposition: HOCHN• → OCHNH•. The last step is the formamide formation: OCHNH• + H• → OCHNH2.
3.1.1 Cluster model
We studied the two first steps of the reaction in the gas phase and studied them again but with one or two water molecules. The reaction profile is represented on Fig. 1. The energies are given with respect to the sum of the energies of HCN and (OH, nH2O) (n = 0,1,2). When we examined the case of the gas phase (n = 0), we observed that the TS2 transition state of hydrogen transposition lies at 11.9 kcalmol-1 above the reference energy. In the interstellar medium, the reaction cannot take place. The addition of water molecules into the reaction has an effect on both transition states. In the first step (OH addition), though the energy of TS1 is lowered, the corresponding barrier energy increases: 5.3 kcalmol-1 (n = 0), 7.7 kcalmol-1 (n = 1), and 9.1 kcalmol-1 (n = 2). This is due to a stabilization between HCN and (OH, nH2O), increasing with n. Even if TS1 decreases with n down to −0.8 kcalmol-1 below the reference for n = 2, the crossing of the barrier does not seem obvious. This would indeed require the use of all the energy of the stabilization of HCN with (OH, 2H2O). But it is more likely that a part of this energy is dissipated in the water ice. The effect of water on the second transition state TS2 is greater than on TS1. Not only does it fall below the reference, but the barrier of the elementary hydrogen transposition step decreases sharply. From 35.9 kcalmol-1 without a water molecule (n = 0), it goes down to 22.6 kcalmol-1 and 19.0 kcalmol-1 for n =1 and n = 2, respectively. Indeed, even 19.0 kcalmol-1 is an impassable barrier in the interstellar medium. But this time, the energy of the transition state TS2 is 14.4 kcalmol-1 below the reference. Even if only some of the energy is recovered by the system from the first step of the reaction path, it is possible to hope to overcome TS2. The results obtained with the cluster model in the previous paragraph unambiguously show the main role of the water: it facilitates the hydrogen transfer. The transition state TS2 of this transfer is then well below the reference energy, and the resulting barrier is hidden. This is not the case for TS1. This reaction start is worth examining using the slab model, which is used to model the solid.
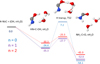 |
Fig. 4 Reaction profiles for HNC + OH→ NHC(•)O (CCSD(T)/cc-pvTZ//B3LYP/cc-pVTZ + ZPE. |
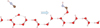 |
Fig. 5 Addition of HNC to OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice. |
3.1.2 Slab model
In this model, we started with a slab on which the OH radical is adsorbed. The position of O from OH adsorbed alone was frozen, while the other atoms of the adsorbate (except C) and O from OH were free. The (HCN,OH) plane was kept unchanged. In the case of path a case, it was done (see Fig. 2) without the formation of the hydrogen bond between the H of HCN and the O of OH. We performed a scan of the C…O distance by steps of 0.1 Å along the elongation axis of the C-O bond of HOHCN by freezing the position of C in each step. In the case of path b, the starting point was HCN interacting with one hydrogen bond. The scan axis was defined by the C of HOCHN (arrival point) and by the C of HCN interacting with the OH with the hydrogen bond (starting point). Path a resulted in a barrier of 4.9 kcalmol-1 and a stabilization of the system of 33.7 kcalmol-1, taking as reference the isolated HCN and OH adsorbed on the H2O slab (see Fig. 3). The reaction energy profile is close to the one obtained by the cluster model (for n = 2, the stabilization is 33.4 kcalmol-1). The barrier, however, is larger, and it should be compared to − 0.8 kcalmol-1 because of the absence of hydrogen bond. This discrepancy can be mainly explained by the difference of the reaction pathway of the two models. Indeed, in the case of the cluster model, the water molecules are free to move, and we did not succeed in forbidding the hydrogen bond between HCN and the water molecules. On the contrary, in the slab model, each water molecule position is constrained by the solid, allowing an HCN attack without involving a hydrogen bond.
In the case of path b in Fig. 2, the barrier increases to 6.2 kcal mol-1 and must be compared to the 9.1 kcal mol-1 value obtained with the cluster model (n = 2). The difference in the barrier between the two models is reasonable when taking into account the use of different functionals and the absence of a basis set superposition error in the case of plane wave basis sets for our slab model. Nevertheless, and most importantly, the conclusion is the same: The C-O bond formation step is not catalyzed by water ice. Finally, a hydrogen atom attacked the adsorbate to obtain the adsorbed formamide (step three). This hydrogenation takes place without barrier, as expected for a radical-radical reaction.
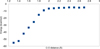 |
Fig. 6 Potential Energy Surface of the addition of HNC on OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice (path a) The reference is the separate fragments. |
3.2 The case of HNC as a reactant
We assumed that the steps are as follows. The first one is the OH addition on HNC: HO• + HNC → HOCNH•. The second one is the H transposition: HOCNH• → OCNH2•. The last one is the formation of formamide: OCNH2• + H• → OCHNH2.
3.2.1 Cluster model
We performed the same modeling with HNC as for HCN. The results are presented in Fig. 4. The difference with HCN is obvious. The formation of the carbon-oxygen bond occurs spontaneously, either in the gas phase (n = 0) or with water molecules (n = 1 or n = 2). From the thermodynamic point of view, the energetic stabilization of the system reaches a value of −30.3 kcal mol-1 in the case n = 2, which is close to that for HCN (−33.4 kcal mol-1). The only transition state is TS2 for the transposition of H. As for HCN, it is clear that water catalyzes this transposition. With a barrier of 41.6 kcalmol-1 and a transition state 7.2 kcalmol-1 above the reference, the hydrogen transposition would be impossible in the gas phase. With water, the barrier drops to 7.6 kcalmol-1 for n =1 and to 4.8 kcalmol-1 for n = 2, respectively 25.5 and 27.7 kcal mol-1 below the reference energy. In these conditions, this barrier is likely to be hidden, allowing the transposition reaction. The tunnel effect would even help.
3.2.2 Slab model
Again, the results obtained with the cluster model in the previous paragraph unambiguously show the role of the water: It facilitates the hydrogen transfer. The energy of this transfer transition state decreases, and the resulting barrier is hidden. The new conclusion in comparison with the case of HCN is that the first addition is barrierless. We decided to confirm this using the slab model (see Fig. 5), which is a more realistic model of the solid. The HNC molecule was placed in the vicinity of the OH radical adsorbed on ice. The conditions of the periodic calculation were identical to those of the HCN case. The addition reaction was studied by performing a scan in which the distance HO… CNH was decreased starting from 2.8 Å until reaching its optimized value in the final product by steps of 0.1 Å. The scan axis is the C-O direction of the adsorbed species. This gave a potential energy surface for the addition of HNC to the adsorbed OH (see Fig. 6). The result showed an absence of barrier, just as with the cluster model. However, there is a difference in the reaction energy. In this case, the energy release due to bond formation is 51.2 kcalmol-1, about 20 kcalmol-1 more than what was obtained with the cluster. While this quantitative difference is significant, the conclusion is the same: The C-O bond forms without a barrier. This additional stabilization provides more energy to get through the H transposition.
4 Conclusions
We studied the formation of formamide from HCN and HNC in damaged water ice by comparing two models. We used cluster models with one or two water molecules as well as slab models taking into account the periodicity of the solid. Despite the very small number of water molecules used to represent the water ice in the cluster models, it is still possible to unambiguously identify trends and draw conclusions. We assumed three steps in the process of formation of formamide: the addition of OH• to HCN or HNC, the transposition of a hydrogen, and finally the addition of a hydrogen. In the first step, in the case of HCN, water ice does not act as a catalyst. Notably, it even increases the energy barrier to form HO-CH=N•. In the case of HNC, there is no barrier anyway, and the only role of water is thus to provide HO radicals. Regarding the second step, it is clear that the transposition of a hydrogen requires the presence of water ice as a catalyst. Finally, the last step is barrier-free, which is not surprising for a reaction between two radicals. We have shown that water ice containing OH provides a possible pathway for the formation of formamide from HNC in the interstellar medium. However, formamide formation seems impossible when starting from HCN.
References
- Barone, V., Latouche, C., Skouteris, D., et al. 2015, MNRAS, 453, L31 [Google Scholar]
- Becke, A. D. 1992, J. Chem. Phys., 96, 2155 [NASA ADS] [CrossRef] [Google Scholar]
- Blöchl, P. E. 1994, Phys. Rev. B, 50, 17953 [CrossRef] [Google Scholar]
- Caricato, M., Frisch, M. J., Hiscocks, J., & Frisch, M. J. 2009, Gaussian 09: IOps Reference (USA: Gaussian Wallingford) [Google Scholar]
- Caselli, P., & Ceccarelli, C. 2012, A&ARv, 20, 1 [CrossRef] [Google Scholar]
- Ceccarelli, C., Caselli, P., Fontani, F., et al. 2017, ApJ, 850, 176 [NASA ADS] [CrossRef] [Google Scholar]
- Chiavassa, T., Borget, F., Aycard, J.-P., Dartois, E., & d’Hendecourt, L. 2005, Actual. Chim., 283, 12 [Google Scholar]
- Darla, N., Sharma, D., & Sitha, S. 2019, J. Phys. Chem. A, 124, 165 [Google Scholar]
- Douglas, K. M., Lucas, D. I., Walsh, C., et al. 2022, ApJ, 937, L16 [CrossRef] [Google Scholar]
- Dulieu, F., Nguyen, T., Congiu, E., Baouche, S., & Taquet, V. 2019, MNRAS, 484, L119 [NASA ADS] [CrossRef] [Google Scholar]
- Ernzerhof, M., & Scuseria, G. E. 1999, J. Chem. Phys., 110, 5029 [NASA ADS] [CrossRef] [Google Scholar]
- Fuente, A., Rodríguez-Franco, A., García-Burillo, S., Martín-Pintado, J., & Black, J. 2003, A&A, 406, 899 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Grimme, S., Antony, J., Ehrlich, S., & Krieg, H. 2010, J. Chem. Phys., 132 [CrossRef] [Google Scholar]
- Herbst, E., & Van Dishoeck, E. F. 2009, ARA&A, 47, 427 [NASA ADS] [CrossRef] [Google Scholar]
- Irvine, W. M., & Schloerb, F. P. 1984, ApJ, 282, 516 [Google Scholar]
- Kresse, G., & Furthmüller, J. 1996, Phys. Rev. B, 54, 11169 [CrossRef] [PubMed] [Google Scholar]
- Kresse, G., & Joubert, D. 1999, Phys. Rev. B, 59, 1758 [CrossRef] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Martínez-Bachs, B., & Rimola, A. 2021, in Computational Science and Its Applications–ICCSA 2021: 21st International Conference, Cagliari, Italy, September 13–16, 2021, Proceedings, Part V 21, 658 [Google Scholar]
- Pérez-Villa, A., Pietrucci, F., & Saitta, A. M. 2020, Phys. Life Rev., 34, 105 [CrossRef] [Google Scholar]
- Puzzarini, C., & Barone, V. 2020, Phys. Life Rev., 32, 59 [NASA ADS] [CrossRef] [Google Scholar]
- Redondo, P., Pauzat, F., Ellinger, Y., & Markovits, A. 2020, A&A, 638, A125 [EDP Sciences] [Google Scholar]
- Redondo, P., Pauzat, F., Markovits, A., & Ellinger, Y. 2021, A&A, 646, A163 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rimola, A., Skouteris, D., Balucani, N., et al. 2018, ACS Earth Space Chem., 2, 720 [NASA ADS] [CrossRef] [Google Scholar]
- Saladino, R., Botta, G., Pino, S., Costanzo, G., & Di Mauro, E. 2012, Chem. Soc. Rev., 41, 5526 [CrossRef] [Google Scholar]
- Schilke, P., Walmsley, C., Pineau Des Forets, G., et al. 1992, A&A, 256, 595 [NASA ADS] [Google Scholar]
- Snyder, L. E., & Buhl, D. 1971, ApJ, 163, L47 [NASA ADS] [CrossRef] [Google Scholar]
- Tennekes, P., Harju, J., Juvela, M., & Tóth, L. 2006, A&A, 456, 1037 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Zuckerman, B., Morris, M., Palmer, P., & Turner, B. 1972, ApJ 173, L125 [Google Scholar]
All Tables
Adsorption energy (kcalmol-1) of HCN on the boat structure surface of hexagonal water ice Ih according to the number of bilayers and the number of bilayers for which the geometry is optimized.
Energy reaction (kcalmol-1) of the HO + HCN = HOHCN reaction calculated using the VASP code and PBE hybrid functionals with variable weights of the exact Hartree-Fock exchange.
All Figures
|
Fig. 1 Reaction profiles for HCN + OH → •NH-C=O (CCSD(T)/cc-pvTZ//B3LYP/cc-pVTZ + ZPE). |
| In the text | |
|
Fig. 2 Attack of HCN on OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice without hydrogen bonding (path a) and with (path b). |
| In the text | |
|
Fig. 3 Potential energy surface of the addition of HCN to OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice (path a). The reference is the separate fragments. |
| In the text | |
|
Fig. 4 Reaction profiles for HNC + OH→ NHC(•)O (CCSD(T)/cc-pvTZ//B3LYP/cc-pVTZ + ZPE. |
| In the text | |
|
Fig. 5 Addition of HNC to OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice. |
| In the text | |
|
Fig. 6 Potential Energy Surface of the addition of HNC on OH adsorbed on the boat surface structure of apolar hexagonal Ih water ice (path a) The reference is the separate fragments. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.