| Issue |
A&A
Volume 644, December 2020
|
|
|---|---|---|
| Article Number | A146 | |
| Number of page(s) | 5 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202039241 | |
| Published online | 11 December 2020 | |
Molecular bending as a vital step toward transforming planar PAHs to fullerenes and tubular structures
1
School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Theoretical Chemistry & Biology, Royal Institute of Technology,
10691
Stockholm,
Sweden
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2
School of Chemistry and Chemical Engineering, Yangzhou University,
225002
Yangzhou,
Jiangsu,
PR China
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
23
August
2020
Accepted:
18
October
2020
Abstract
Context. Polycyclic aromatic hydrocarbons (PAHs) and fullerenes are the largest molecules found in the interstellar medium (ISM). They are abundant and widespread in various astronomical environments. However, the detailed connection between these two species is unknown; in particular, no quantum chemical studies have been performed.
Aims. In this work, we investigate a vital step in transforming planar PAHs to fullerenes, that is, the tubulation processes of PAHs.
Methods. We used density functional theory for this study. The molecular structures and vibrational frequencies were calculated using the hybrid density functional B3LYP method. To better describe intermolecular forces, we considered Grimme’s dispersion correction in the calculations for this work. Intrinsic reaction coordinate calculations were also performed to confirm that the transition state structures are connected to their corresponding local potential energy surface minima.
Results. As expected, we find that it is easier to bend a molecule as it gets longer, whereas it is harder to bend the molecule if it gets “wider” (i.e., with more rows of benzene rings). The change of multiplicity slightly alters the bending energies, while (a complete) dehydrogenation alleviates the bending barrier significantly and facilitates the formation of pentagons, which may act as an indispensable step in the formation of fullerenes in the ISM.
Key words: astrochemistry / molecular data / molecular processes / infrared: ISM / ISM: molecules
© ESO 2020
1 Introduction
Polycyclic aromatic hydrocarbon (PAH) molecules, as revealed by the distinctive set of emission bands at 3.3, 6.2, 7.7, 8.6, 11.3, and 12.7 μm characteristic of their vibrational modes, are abundant and widespread throughout the Universe (Leger & Puget 1984; Allamandola et al. 1985; Henning & Salama 1998; Tielens 2008). These molecules lock up ≲15% of the interstellar carbon and their emission accounts for up to 20% of the total infrared (IR) power of the Milky Way and star-forming galaxies (Li 2020). In the absence of steric repulsions, the sp2 hybridized orbitals of PAHs give rise to an extremely stable and fully conjugated delocalized aromatic π-electron system, which supports a planar structure. Density functional theory (DFT) has predicted that equilibrium geometries show planarity for large compact PAHs up to C384H48 (Ricca et al. 2012). Nevertheless, single graphene sheets, which may be considered as an extended dehydrogenated PAHs, are expected to bend, implying that planar structures are unstable with respect to the formation of curved structures such as soot, fullerenes, and nanotubes (Novoselov et al. 2004).
Chuvilin et al. (2010) experimentally found a transformation from a graphene sheet to a fullerene following the loss of carbon atoms at the edge of graphene and molecular curving using an aberration-corrected transmission electron microscopy. Zhen et al. (2014) showed that fullerenes can also be formed from large PAHs directly through photodissociation experiments. On the other hand, theoretical studies have exhibited molecular curling processes of linear PAHs and graphitic ribbons (Robertson et al. 1992; Pietrucci & Andreoni 2014; Segawa et al. 2016; Chen et al. 2018; Chen & Luo 2019). In particular, Robertson et al. (1992) found that larger ribbons (e.g., C108) may bend to an open-ended hollow carbon structure representing a seemingly fullerene precursor. Pietrucci & Andreoni (2014) showed a clear reaction route for the formation of a fullerene from a graphene flake using ab initio molecular dynamics simulations. On the other hand, C60 and its cations are ubiquitously seen in the interstellar and circumstellar medium through their characteristic vibrational spectral bands in the infrared and the diffuse bands of C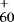 close to 1 μm, which are believed to be formed from large PAHs through photo-induced top-down reactions (Cami et al. 2010; Sellgren et al. 2010; Berné et al. 2013, 2015; Strelnikov et al. 2015).
close to 1 μm, which are believed to be formed from large PAHs through photo-induced top-down reactions (Cami et al. 2010; Sellgren et al. 2010; Berné et al. 2013, 2015; Strelnikov et al. 2015).
Recently, we have shown that a closed steric hydrocarbon nanostructure could be formed in interstellar space from benzene and phenyl through molecular bending (Chen & Li 2019). This formation pathway starts with a benzene combining with a phenyl radical (C6 H5•) to form a biphenyl radical (C12 H10•) through hydrogen abstraction. Then, the biphenyl radical dissociates a hydrogen atom and reacts with another phenyl radical to form a triphenyl radical (C18 H14•). The triphenyl radical, with the loss of a hydrogen atom, isomerizes to a closed 3D structure. Subsequently, the conventional hydrogen-abstraction and acetylene-addition (HACA; Frenklach & Feigelson 1989) process, which involves repetitive hydrogen losses from the 3D structure followed by the addition of one or two acetylene (C2 H2) molecule(s), leads to the continual growth of the 3D structure to more complex armchair carbon nanotubes (Chen & Li 2019).
This work extends previous studies to a general scenario of molecular bending or curling. Using first-principles methods, we investigate the influence of geometry, hydrogenation, and charge state on the bending processes of PAHs. A clear picture is drawn for the connections among PAHs, nanotubes, and fullerenes. This work provides important information for the detection of new carbonaceous species in space. In addition, it also enable us to understand the origin, formation, and evolution of organic molecules inthe interstellar medium (ISM).
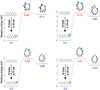 |
Fig. 1 Calculated dissociation energies and molecular bending barriers of tetracene (C18 H12), pentacene (C22H14), hexacene (C26H16), and heptacene (C30H18). The dissociation energy for removing a terminal side hydrogen atom from these molecules is about 4.8 eV. The molecular bending barrier decreases from ~6.7 to ~4.2 eV as the length of the molecule increases. Following the transition states (shown in red), closed hollow carbon structures can be formed. The blue circles highlight the region where the reactions take place. |
2 Methods
Apparently, molecular bending is critical to the formation of soot, fullerenes, and nanotubes (Curl & Smalley 1988; Robertson et al. 1992; Berné & Tielens 2012; Chen & Li 2019). To this end, in this work we performed a systematic DFT study on the bending process of PAHs via the Gaussian16 package (Frisch et al. 2016). All structures were optimized using the hybrid density functional B3LYP method (Lee et al. 1988; Becke 1992) with the 6-311++G(2d,p) basis set. To take the intermolecular van der Waals forcesinto account, we included the D3 version of Grimme’s correction with Becke-Johnson damping (Grimme et al. 2011) in the calculations. The vibrational frequencies were calculated for the optimized geometries to verify that these correspond to minima or first-order saddle points (transition states) on the potential energy surface (PES). We took the zero point vibrational energies (ZPVE) into account. Intrinsic reaction coordinate (IRC) calculations (Fukui 1981; Dykstra 2005) were performed to confirm that the transition state structures are connected to their corresponding local PES minima.
3 Results and discussions
Figure 1 shows the general reaction path (including structures and energies) for the bending of four linear PAHs: tetracene (C18 H12), pentacene (C22 H14), hexacene (C26 H16), and heptacene (C30 H18). At first, a terminal side hydrogen atom was removed from the molecule. Such an H loss is a barrier-free reaction (Holm et al. 2011; Chen et al. 2015), and it is a key step in molecular bending, which leads to the formation of a dangling bond. Thereby, the molecules may bend to a curved structure and a closed 3D structure correspondingly by overcoming various reaction barriers, depending on the length of the specific molecule. It is found that the bending barriers for tetracene (~6.7 eV) are rather high in comparison to dissociation channels, for example, ~5 eV for H, H2, and C2 H2 losses (Holm et al. 2011; Chen et al. 2015). Therefore, such short molecules ought to be dissociated before any bending occurs. As the length of the molecule increases, the bending barrier decreases, for example, to ~4.9 eV for hexacene and to ~4.2 eV to heptacene, which is already lower than the H, H2, and C2 H2 loss energies. Furthermore, we also note that the strength (measured as the energy between the product and the transition state) of the closed 3D structures increases as the molecules get longer. For tetracene, the strength is only about 0.3 eV, while it increases to about 1 eV for hexacene and heptacene. Segawa et al. (2016) derived the strain energy of carbon nanotubes and nanobelts as a function of the repeat unit. We compared our results with the values derived from their functions and find that the two values have the same order of magnitude. The difference between barriers for molecules such as hexacene and heptacene is ~0.7 eV; the corresponding difference in strain energy is ~0.2 eV.
We wondered what happens with the bending when the molecule gets wider. Figure 2 shows how the bending energy evolves as the PAH width (i.e., the number of benzene rows) increases. The molecules selected for this comparison include C31 H15 (a two-row molecule), C40 H16 (a three-row molecule), and circumovalene (C66 H20, a five-row molecule) bending in two different directions. We find that the bending energy increases as the molecule grows wider (i.e., the number of benzene rows increases). For circumovalene, two bending pathways are studied, depending on the bending directions. The calculations show that it is harder to bend the molecule in the direction perpendicular toward the C–C or C =C bonds (~16.3 eV) than in the direction parallel toward the C–C or C =C bonds (~13.9 eV), with an energy difference of ~2.4 eV.
Besides a single H loss, the H2 loss is also a dominant dissociation pathway for PAHs, in which the lowest barriers come from the H migration to a neighboring carbon on the same ring and the highest transition state for H2 loss is about 5 eV (Paris et al. 2014; Chen et al. 2015). We have, therefore, investigated the bending barriers following an H2 loss from PAHs. Figure 3 demonstrates the bending processes for neutral heptacene with singlet and triplet multiplicities. Two transition states are found for the double dehydrogenated heptacene. The first barrier is about 4.7 eV for the original multiplicity of M1 and ~4.4 eV for the original multiplicity of M3, while the second barrier is ~5.7 eV for the original multiplicity of M1 and goes down to ~5.4 eV for the original multiplicity of M3. Similar reaction pathways and barriers are found for the singly-charged systems, that is, the energies andstructures do not significantly change with a different charge state or a different multiplicity. See Fig. 3 for the details of various reaction pathways.
It has been suggested that fullerenes can be formed from large PAHs following fully dehydrogenations (Berné & Tielens 2012; Zhen et al. 2014; Pietrucci & Andreoni 2014). We find that following the dehydrogenations, the bending barriers decreased significantly, from ~13.9 to ~8.2 eV (see Figs. 2 and 4). Upon overcoming the barrier, two covalent bonds and a pentagon can be formed, which is crucial to producing fullerenes. In addition, such an energy (~8.2 eV) can be easily accessed in the ISM following the absorption of a single UV photon, which is consistent with the hypothesis that fullerenes can be formed from large PAHs in the ISM (Berné & Tielens 2012; Zhen et al. 2014).
 |
Fig. 2 Calculated dissociation energies and molecular bending barriers of pentacene (a linear molecule), C31 H15 (a two-row molecule), C40H16 (a three-row molecule), and circumovalene. The dissociation energy for removing a terminal side hydrogen atom from these molecules is about 4.8 eV. The molecular bending barrier increases from ~5.5 to ~13.9 eV as the number of rows increases. For ovalene, two different bending pathways, depending on the position of H loss (denoted with a yellow circle), are investigated. The barrier increases from ~13.9 eV for the loss of a duo-hydrogen to ~16.3 eV for the loss of a solo-hydrogen. |
 |
Fig. 3 Reaction pathways and energies for molecular bending of a neutral heptacene following a H2 loss. The table on the right shows the energies for different multiplicities and charges. |
 |
Fig. 4 Formation of a pentagon following molecular bending of a fully dehydrogenated compact PAH (graphene sheet). The formed pentagon is highlighted in blue. Subsequently, the molecule might convert to a fullerene at high temperatures following C2 losses and exhibit further curving using the ab initio molecular dynamics simulations (Pietrucci & Andreoni 2014). |
4 Summary
In this work, we have shown that molecular bending is closely associated with the size and shape of the molecule. As expected, we find that fora fixed width, the bending energy decreases as the length of the molecule increases. While for a fixed length (± a benzene-ring), the bending energy increases with the molecular height. Moreover, the bending energy also depends on the bending directions; for example, for the direction perpendicular to most of the C–C or C =C bonds, the bending energy is higher than the direction parallel to most of the C–C or C =C bonds. For circumovalene, the energy difference for the two extreme directions is about 2.4 eV.
As of the influence of multiplicities on molecular bending, the change of multiplicities slightly alter the bending energies by about 0.3 eV. In addition, wenote that the bending energy increases with a double hydrogen loss in comparison to a single hydrogen loss. However, following the complete loss of all the hydrogens, a compact fully dehydrogenated PAH, circumovalene reduces its bending energy to ~8.2 eV. This value is lower than its precursor, that is, circumovalene with a single H loss, and much lower than the Lyman limit (13.6 eV). More importantly, a pentagon can be formed following the molecular bending. Such a process ought to be a crucial and indispensable step for the formation of fullerenes in space.
Acknowledgements
The calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC). Y.W. acknowledges the Thousand Talents Plan for Young Professionals of China.
References
- Allamandola, L., Tielens, A. G. G. M., & Barker, J. 1985, ApJ, 290, L25 [NASA ADS] [CrossRef] [Google Scholar]
- Becke, A. D. 1992, J. Chem. Phys., 96, 2155 [NASA ADS] [CrossRef] [Google Scholar]
- Berné, O., & Tielens, A. G. 2012, Proc. Natl. Acad. Sci., 109, 401 [NASA ADS] [CrossRef] [Google Scholar]
- Berne, O., Mulas, G., & Joblin, C. 2013, A&A, 550, L4 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Berné, O., Montillaud, J., & Joblin, C. 2015, A&A, 577, A133 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cami, J., Bernard-Salas, J., Peeters, E., & Malek, S. E. 2010, Science, 329, 1180 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Chen, T., & Li, A. 2019, A&A, 631, A54 [CrossRef] [EDP Sciences] [Google Scholar]
- Chen, T., & Luo, Y. 2019, MNRAS, 486, 1875 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T., Gatchell, M., Stockett, M. H., et al. 2015, J. Chem. Phys., 142, 144305 [NASA ADS] [CrossRef] [Google Scholar]
- Chen, T., Zhen, J., Wang, Y., Linnartz, H., & Tielens, A. G. G. M. 2018, Chem. Phys. Lett., 692, 298 [NASA ADS] [CrossRef] [Google Scholar]
- Chuvilin, A., Kaiser, U., Bichoutskaia, E., Besley, N. A., & Khlobystov, A. N. 2010, Nat. Chem., 2, 450 [NASA ADS] [CrossRef] [Google Scholar]
- Curl, R. F., & Smalley, R. E. 1988, Science, 242, 1017 [CrossRef] [Google Scholar]
- Dykstra, C. E. 2005, Theory and Applications of Computational Chemistry: The First Forty Years (Amsterdam: Elsevier) [Google Scholar]
- Frenklach, M., & Feigelson, E. D. 1989, ApJ, 341, 372 [NASA ADS] [CrossRef] [Google Scholar]
- Frisch, M., Trucks, G., Schlegel, H., et al. 2016, Gaussian 16, Revision C.01 (Wallingford CT) [Google Scholar]
- Fukui, K. 1981, Acc. Chem. Res., 14, 363 [CrossRef] [Google Scholar]
- Grimme, S., Ehrlich, S., & Goerigk, L. 2011, J. Comput. Chem., 32, 1456 [Google Scholar]
- Henning, T., & Salama, F. 1998, Science, 282, 2204 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Holm, A. I., Johansson, H. A., Cederquist, H., & Zettergren, H. 2011, J. Chem. Phys., 134, 044301 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Leger, A., & Puget, J. 1984, A&A, 137, L5 [NASA ADS] [Google Scholar]
- Li, A. 2020, Nat. Astron., 4, 339 [CrossRef] [Google Scholar]
- Novoselov, K. S., Geim, A. K., Morozov, S. V., et al. 2004, Science, 306, 666 [CrossRef] [PubMed] [Google Scholar]
- Paris, C., Alcamí, M., Martín, F., & Díaz-Tendero, S. 2014, J. Chem. Phys., 140, 204307 [NASA ADS] [CrossRef] [Google Scholar]
- Pietrucci, F., & Andreoni, W. 2014, J. Chem. Theory Comput., 10, 913 [CrossRef] [Google Scholar]
- Ricca, A., Bauschlicher Jr. C. W., Boersma, C., Tielens, A. G., & Allamandola, L. J. 2012, ApJ, 754, 75 [NASA ADS] [CrossRef] [Google Scholar]
- Robertson, D., Brenner, D., & White, C. 1992, J. Phys. Chem., 96, 6133 [CrossRef] [Google Scholar]
- Segawa, Y., Yagi, A., Ito, H., & Itami, K. 2016, Org. Lett., 18, 1430 [CrossRef] [Google Scholar]
- Sellgren, K., Werner, M. W., Ingalls, J. G., et al. 2010, ApJ, 722, L54 [NASA ADS] [CrossRef] [Google Scholar]
- Strelnikov, D., Kern, B., & Kappes, M. 2015, A&A, 584, A55 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Tielens, A. G. G. M. 2008, ARA&A, 46, 289 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Zhen, J., Castellanos, P., Paardekooper, D. M., Linnartz, H., & Tielens, A. G. 2014, ApJ, 797, L30 [NASA ADS] [CrossRef] [Google Scholar]
All Figures
|
Fig. 1 Calculated dissociation energies and molecular bending barriers of tetracene (C18 H12), pentacene (C22H14), hexacene (C26H16), and heptacene (C30H18). The dissociation energy for removing a terminal side hydrogen atom from these molecules is about 4.8 eV. The molecular bending barrier decreases from ~6.7 to ~4.2 eV as the length of the molecule increases. Following the transition states (shown in red), closed hollow carbon structures can be formed. The blue circles highlight the region where the reactions take place. |
| In the text | |
|
Fig. 2 Calculated dissociation energies and molecular bending barriers of pentacene (a linear molecule), C31 H15 (a two-row molecule), C40H16 (a three-row molecule), and circumovalene. The dissociation energy for removing a terminal side hydrogen atom from these molecules is about 4.8 eV. The molecular bending barrier increases from ~5.5 to ~13.9 eV as the number of rows increases. For ovalene, two different bending pathways, depending on the position of H loss (denoted with a yellow circle), are investigated. The barrier increases from ~13.9 eV for the loss of a duo-hydrogen to ~16.3 eV for the loss of a solo-hydrogen. |
| In the text | |
|
Fig. 3 Reaction pathways and energies for molecular bending of a neutral heptacene following a H2 loss. The table on the right shows the energies for different multiplicities and charges. |
| In the text | |
|
Fig. 4 Formation of a pentagon following molecular bending of a fully dehydrogenated compact PAH (graphene sheet). The formed pentagon is highlighted in blue. Subsequently, the molecule might convert to a fullerene at high temperatures following C2 losses and exhibit further curving using the ab initio molecular dynamics simulations (Pietrucci & Andreoni 2014). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.