| Issue |
A&A
Volume 644, December 2020
|
|
|---|---|---|
| Article Number | A3 | |
| Number of page(s) | 10 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202038766 | |
| Published online | 24 November 2020 | |
Formation of interstellar cyanoacetamide: a rotational and computational study★
1
Grupo de Espectroscopía Molecular (GEM), Edificio Quifima, Área de Química-Física, Laboratorios de Espectroscopía y Bioespectroscopía, Parque Científico UVa, Unidad Asociada CSIC,
47011
Valladolid, Spain
2
Computational Chemistry Group, Departamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid,
47011
Valladolid, Spain
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
26
June
2020
Accepted:
22
September
2020
Abstract
Context. Cyanoacetamide is a –CN bearing molecule that is also an amide derivative target molecule in the interstellar medium.
Aims. The aim of our investigation is to analyze the feasibility of a plausible formation process of protonated cyanoacetamide under interstellar conditions and to provide direct experimental frequencies of the ground vibrational state of the neutral form in the microwave region in order to enable its eventual identification in the interstellar medium.
Methods. We used high-level theoretical computations to study the formation process of protonated cyanoacetamide. Furthermore, we employed a high-resolution laser-ablation molecular beam Fourier transform spectroscopic technique to measure the frequencies of the neutral form.
Results. We report the first rotational characterization of cyanoacetamide, and a precise set of the relevant rotational spectroscopic constants have been determined as a first step to identifying the molecule in the interstellar medium. We fully explored the potential energy surface to study a gas-phase reaction on the formation process of protonated cyanoacetamide. We found that an exothermic process with no net activation barrier is initiated by the high-energy isomer of protonated hydroxylamine, which leads to protonated cyanoacetamide.
Key words: molecular data / ISM: molecules / astrochemistry / molecular processes
The catalog of lines is only available at the CDS via anonymous ftp to cdsarc.u-strasbg.fr (130.79.128.5) or via http://cdsarc.u-strasbg.fr/viz-bin/cat/J/A+A/644/A3
© ESO 2020
1 Introduction
More than 30 molecules containing the cyano (–C ≡N) functional group have been observed to date in diverse types of astronomical environments. Among these are the well-known family of cyanopolyynes, H–(C ≡C)n–C ≡N, which are commonly found in cold interstellar clouds (Broten et al. 1978; Bell et al. 1997), in exoplanetary envelopes, such as the carbon- and nitrogen-rich atmosphere of Titan (Kunde et al. 1981; Coustenis et al. 1991), and in circumstellar envelopes around the stars (Winnewisser & Walmsley 1978; Pardo et al. 2005). In addition, dicyanopolyynes, defined as molecules that contain two cyano groups, N ≡C–(C ≡C)n–C ≡N, could also be abundant in interstellar and circumstellar clouds (Kołos & Grabowski 2000; Petrie et al. 2003). Very recently, isocyanogen (CNCN), the simplest member of this series, has been detected in the interstellar medium (ISM; Agúndez et al. 2018). However, many simple nitriles have yet to be discovered.
The symbiotic relationship between astronomical searches and laboratory experiments using rotational spectroscopy has allowed successfully identifying many interstellar compounds (Herbst & van Dishoeck 2009). In 2008, aminoacetonitrile (NH2CH2CN) was discovered in Sgr B2(N) (Belloche et al. 2008) by means of rotational spectroscopy data (Bogey et al. 1990). Kolesniková et al. (2017a) carried out a detailed analysis of its rotational spectra up to 480 GHz. Furthermore, acetamide (CH3C(O)NH2), and N-metylformamide (HC(O)NHCH3), two N-bearing molecules containing a peptide-type bond (–NH–C(O)–), have also been detected in space (Hollis et al. 2006; Belloche et al. 2017). These molecules are very interesting from a prebiotic point of view because the amide linkage is an essential feature in proteins. In current astronomical research, great efforts have been made to study more complex members of these molecular systems, such as glycolamide (CH2(OH)C(O)NH2) (Sanz-Novo et al. 2020).
The astronomical detection of isopropyl cyanide ((CH3)2CHCN), the first branched hydrocarbon detected in the ISM (Belloche et al. 2014), was of paramount importance. Recently, a comprehensive analysis of its millimeter- and submillimeter-wave spectra has been reported (Kolesniková et al. 2017b). Observations of more branched-chain molecules in the ISM will firmly establish the link between the chemical composition of the ISM and the chemical molecular of small bodies such as comets and asteroids in our Solar System, in which there is a dominance of branched-chain amino acids as compared to straight chains. This suggests that cyanoacetamide (CH2(CN)C(O)NH2), an amide derivative, and also a branched-chain –CN containing molecule, may exist in the ISM and motivate astronomers to search for it.
Cyanoacetamide is a complex organic molecule (COM) from an astronomical point of view (Herbst & van Dishoeck 2009). This molecule is of great importance in organic chemistry and biology; it assumes a special role in heterocyclic chemistry (El Bialy et al. 2011). It is a solid in standard conditions for temperature and pressure, with a melting point of 119–121 °C. So far, no detailed experimental structural information of the isolated molecule has been reported. Only an assignment of its IR and Raman spectra has been reported quite a long time ago by Haverbeke & Herman (1975), but its conformational panorama remains unknown. Fourier transform (FT) microwave spectroscopy is extremely sensitive to molecular geometry and has been proved a powerful tool in the conformational analysis (Schermann 2008). Coupled with laser-ablation techniques, it enables an ultra-fast vaporization of organic and biological molecules and allows them to be probed in the isolated environment of a supersonic expansion (Alonso et al. 2014). With this outstanding strategy, several relevant biomolecules and most of the amino acids (Alonso et al. 2015), as well as species of astrophysical interest, such as hydantoin (Alonso et al. 2017) and glycinamide (Alonso et al. 2018), have recently been revealed.
Moreover, new gas-phase theoretical and experimental studies exploring different nitrile species should represent an essential source of reference data in constraining models of nitrile formation in the ISM and in predicting which other nitrile species might be present and detectable. The knowledge of their formation processes is essential to understand the evolution of more complex molecules. The chemical diversity and complexity of astronomical sources could be explained as the result of gas, grain, and gas-grain interactions in dense interstellar clouds. The study of surface processes has recently attracted the attention of many theoretical and experimental researchers as an alternative to gas-phase reactions and also to understand the compositionof icy mantles. Some interstellar molecules are formed from dust-grain mantles. In this context, it was concluded in several experimental studies (He et al. 2015; Zheng & Kaiser 2010; Congiu et al. 2012) that hydroxylamine, which is expected to be one of the main precursors of amino acids in space, could be generated in the solid phase and then desorbed through a temperature-induced process into the gas phase. However, exothermic ion-molecule reactions between positive ions and neutral molecules still play a crucial role in the synthesis of organic interstellar molecules in the gas phase (Barrientos et al. 2012; Redondo et al. 2014, 2015, 2017) because these processes tend to have negligible activation energy barriers. Thus, we have carried out an accompanying theoretical study, consisting of a full exploration of the potential energy surface (PES), to analyze the feasibility of a plausible gas-phase reaction as a source of protonated cyanoacetamide in the ISM and thereby disclose the corresponding neutral form as a candidate molecule to be searched for in the ISM. This is reported in Sect. 3.1.
Following our previous work (Sanz-Novo et al. 2020), we propose the study of cyanoacetamide, another amide derivative, as a candidate molecular system for interstellar detection. In order to help in its laboratory or astronomical detection, the main aim of this study is to provide direct experimental information of the ground vibrational state of cyanoacetamide in the microwave frequency region as the first step of the astrophysical search. In this work, the experimental values of the 14N nuclear quadrupole coupling constants together with the rotational constants are provided in order to perfectly reproduce the spectrum. For observations in low-frequency regions such as those available for the Green Bank Telescope (GBT) or the Effelsberg 100 m Radio Telescope, the hyperfine structure could be a drawback for interpreting the astronomical spectra. The Effelsberg observatory, for instance, has the capability to search for transitions in the microwave region such as the lines we aim to provide in this work (Sanz-Novo et al. 2020). For some interstellar molecules such as cyanoallene and methanimine, the interpretation of the hyperfine pattern was moreover crucial for its conclusive identification in the ISM (Lovas et al. 2006; Godfrey et al. 1973). This motivated us to record the microwave rotational spectrum of cyanoacetamide using the high-resolution laser-ablation molecular beam Fourier transform (LA-MB-FTMW) spectrometer at the University of Valladolid. In addition, we have carried out a theoretical study of the gas-phase formation process of protonated cyanoacetamide from the reaction of protonated hydroxylamine and cyanoacetaldehyde. To the best of our knowledge, no theoretical studies have been devoted to proposing potential mechanisms of cyanoacetamide formation in the ISM. The astrochemical interpretation of these might pave the route to the elucidation of models of nitrile-derivatives formation in space and may act as a source of reference data to reveal the evolution of the more complex molecules.
2 Methods
2.1 Computational section
The structures of all the species studied in this work (reactants, intermediates, transition states, and products) were first optimized in the framework of the density functional theory (DFT) employing the B3LYP exchange-correlation functional, which includes the Becke’s three-parameter exchange functional (Becke 1993) and the Lee–Yang–Parr correlation functional (Lee et al. 1988). In addition, Grimme’s D3 dispersion interactions were included (Grimme et al. 2010). The correlation-consistent polarized valence triple-ζ, aug-cc-pVTZ basis set including diffuse functions was used in these computations (Dunning 1989; Woon & Dunning 1993). Subsequently, the structures were reoptimized at the MP2 level (second-order Møller-Plesset perturbation theory; Møller & Plesset 1934) and using a double-hybrid density functional (B2PLYPD) with long-range dispersion interactions (Schwabe & Grimme 2007). At the respective optimized geometries, harmonic vibrational frequencies were calculated to apply zero-point-energy corrections to the electronic energy. Moreover, this allowed us to verify the nature of the stationary points located on the PES, either a transition state (TS) with one of the frequencies, and just one imaginary or a true minimum with all vibrational frequencies real. The intrinsic reaction coordinate (IRC) technique (Gonzalez & Schlegel 1991) was used to confirm the connections between TS structures and adjacent minima. We have also computed anharmonic vibrational frequencies for the most stable conformer of cyanoacetamide at the MP2 level using a second-order perturbation treatment (VPT2; Hoy et al. 1972), which includes a full cubic force field (CFF) and semidiagonal quartic force constants. Finally, in order to compute more accurate energies, we carried out coupled-cluster computations. Specifically, the coupled-cluster single- and double-excitation model augmented with a noniterative triple-excitation correction, CCSD(T) (Raghavachari et al. 1989) coupled with the aug-cc-pVTZ basis set on the B2PLYPD3/aug-cc-pVTZ optimized geometries was considered. All quantum-mechanical calculations were carried out with the Gaussian 16 program package (Gaussian 16 Revision A.03).
2.2 Experimental section: LA-MB-FTMW spectroscopy
A commercial sample of cyanoacetamide was purchased from Sigma Aldrich and used without further purification (99%). It is a white crystalline substance with a melting point of 119–121 °C. The rotational spectrum was recorded using a LA-MB-FTMW spectrometer, which works in the frequency range 8–18 GHz and has been described elsewhere (Alonso et al. 2009; Bermudez et al. 2014). Using this technique, we were able to provide rotational signatures of cyanoacetamide to unravel its conformational preferences and fully resolve its 14N hyperfine structure. The laser-ablated products were seeded in Ne (stagnation pressure 10 bar) and adiabatically expanded to form a supersonic jet into a Fabry–Pérot resonator. A short microwave radiation pulse (0.3 μs) was then applied to polarize the vaporized molecules macroscopically. A microwave radiation pulse sequence that allowed us to collect multiple free-induction decay (FID) has recently been implemented (León et al. 2017). The microwave FID was recorded in the time domain and Fourier transformed to the frequency domain. The pulsed molecular beam was introduced parallel to the axis of the resonator, so that each observed transition appears as a Doppler doublet. The resonance frequency is then calculated by the arithmetic mean of the two Doppler components.
3 Results and discussion
3.1 Formation process of protonated cyanoacetamide
In a first step of the investigation, we studied the gas-phase formation process of protonated cyanoacetamide [C3 N2H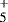 ] from the reaction of protonated hydroxylamine and cyanoacetaldehyde (CNCH2C(O)H). We therefore carried out a full exploration of the singlet potential energy surfaces because all the reactants are in their singlet electronic state (1A’). Our main goal in this section is to analyze whether the reactions we studied might be a source of protonated cyanoacetamide, and we discuss the results in the context of the ISM. In conditions of the ISM, basically low temperatures and low densities, interstellar viable reactions should be exothermic and barrier-free processes. Only a few protonated complex organic molecules (COMs) have been detected in the ISM so far, although it seems that they could evolve to the corresponding neutral species by dissociative recombination.
] from the reaction of protonated hydroxylamine and cyanoacetaldehyde (CNCH2C(O)H). We therefore carried out a full exploration of the singlet potential energy surfaces because all the reactants are in their singlet electronic state (1A’). Our main goal in this section is to analyze whether the reactions we studied might be a source of protonated cyanoacetamide, and we discuss the results in the context of the ISM. In conditions of the ISM, basically low temperatures and low densities, interstellar viable reactions should be exothermic and barrier-free processes. Only a few protonated complex organic molecules (COMs) have been detected in the ISM so far, although it seems that they could evolve to the corresponding neutral species by dissociative recombination.
According to Snow et al. (2007), the two isomers of protonated hydroxylamine (NH3 OH+ and NH2 OH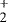 ) might be obtained in the protonation process of hydroxylamine using CH
) might be obtained in the protonation process of hydroxylamine using CH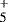 as protonating agent. Neither of them reacts with H2 under interstellar conditions (Largo et al. 2009), and if they exist in the ISM, they may therefore be able to react with other molecules, for instance, with cyanoacetaldehyde.
as protonating agent. Neither of them reacts with H2 under interstellar conditions (Largo et al. 2009), and if they exist in the ISM, they may therefore be able to react with other molecules, for instance, with cyanoacetaldehyde.
Neither protonated hydroxylamine nor cyanoacetaldehyde have been detected in space to date. However, hydroxylamine was produced byultraviolet laser irradiation of ice mixtures simulating interstellar grains (Nishi et al. 1984). In addition, formation of hydroxylamine was observed in electron-irradiated ammonia–water ices at temperatures of 10 and 50 K (Zheng & Kaiser 2010) and also in electron-irradiated ammonia-oxygen ices at astrophysically relevant temperatures of 5.5 K (Tsegaw et al. 2017). Hydroxylamine is usually taken as reactant in the synthesis of possible interstellar molecules (Barrientos et al. 2012).
The rotational spectrum and conformational composition of cyanoacetaldehyde has previously been reported by Møllendal et al. (2012). Its existence in interstellar space and in planetary atmospheres is quite likely because of the facile formation of cyanoacetylene in liquid water (Ferris et al. 1968), presumably formed in a catalytic process (Horn et al. 2008). It is known that cyanoacetylene, a compound formed in a spark-discharge reaction in a mixture of methane and nitrogen (Sánchez et al. 1967), is widespread in the ISM, and found in comets (Bockelée-Morvan et al. 2000) and in the atmosphere of Titan (Coustenis et al. 1999). However, several gas-phase models of an uncatalyzed addition reaction between the two neutral molecules cyanoacetylene and water as well as the addition of water to protonated cyanoacetylene, which also exists in the ISM (Kawaguchi et al. 1994), followed by an electron recombination reaction have been predicted to be inefficient by quantum-chemical calculations (Horn et al. 2008). Furthermore, we studied a plausible formation route showing that cyanoacetaldehyde could be formed by the addition of the O (3P) radical to vinyl cyanide, which is a well-known interstellar molecule (for detailed information, see Fig. B.1).
The reactions between protonated hydroxylamine (NH3 OH+/ NH2 OH2 +) and cyanoacetaldehyde to give protonated cyanoacetamide can be summarized as follows:
![Mathematical equation: \begin{equation*} \textrm{NH}_3\textrm{OH}^+ + \textrm{CNCH}_2\textrm{C(O)H} \rightarrow [\textrm{C}_3\textrm{N}_2{\textrm{H}_5^+}] + \textrm{H}_2\textrm{O}\end{equation*}](/articles/aa/full_html/2020/12/aa38766-20/aa38766-20-eq4.png) (1)
(1)
![Mathematical equation: \begin{equation*} \textrm{NH}_2{\textrm{OH}_2^+} + \textrm{CNCH}_2\textrm{C(O)H} \rightarrow [\textrm{C}_3\textrm{N}_2{\textrm{H}_5^+}] + \textrm{H}_2\textrm{O}. \end{equation*}](/articles/aa/full_html/2020/12/aa38766-20/aa38766-20-eq5.png) (2)
(2)
Protonation of cyanoacetamide gives two isomers with the [C3 N2H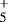 ] molecular formula: O-protonated cyanoacetamide, (O-prot) CNCH2COHNH
] molecular formula: O-protonated cyanoacetamide, (O-prot) CNCH2COHNH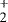 , and N-protonated cyanoacetamide, CNCH2C(O)NH
, and N-protonated cyanoacetamide, CNCH2C(O)NH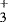 . Protonationon the oxygen atom is the most favourable process because (O-prot) CNCH2COHNH
. Protonationon the oxygen atom is the most favourable process because (O-prot) CNCH2COHNH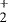 is located 12.2 kcal mol−1 lower in energy, at the CCSD(T) level, than the corresponding N-protonated isomer.
is located 12.2 kcal mol−1 lower in energy, at the CCSD(T) level, than the corresponding N-protonated isomer.
In Table1, we collect the relative energies with respect to reactants of the possible products that can be formed in the reaction between either NH3 OH+ or NH2 OH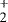 and CNCH2C(O)H computed at different levels of theory. At the CCSD(T) level of theory, hydroxylamine protonated in its oxygen atom, NH2 OH
and CNCH2C(O)H computed at different levels of theory. At the CCSD(T) level of theory, hydroxylamine protonated in its oxygen atom, NH2 OH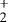 , was found to lie 25.5 kcal mol−1 higher in energy than the most stable isomer, NH3 OH+. This value therefore is the difference of the reaction energies of paths 1 (first entry of Table 1) and 2 (second entry) computed at this level of theory.
, was found to lie 25.5 kcal mol−1 higher in energy than the most stable isomer, NH3 OH+. This value therefore is the difference of the reaction energies of paths 1 (first entry of Table 1) and 2 (second entry) computed at this level of theory.
Table 1 shows that all reactions we studied are exothermic processes. Regardless of the level of theory, the most favourable process from thermodynamic arguments is the formation of O-protonated cyanoacetamide. This might have been expected, and it mainly arises from the different chemical natures of the isomers. The multiple topological dispositions of the hydrogen atom allow us to generate different arrangements of the –NH and –OH groups. This leads to the formation ofspecific intramolecular hydrogen bonds that further stabilize this molecular system. In particular, the N–H⋯π weak hydrogen bond is expected to be one of the leading structural motifs stabilizing the structure, as pointed out in previous studies (Cabezas et al. 2012).
We focused on the reactions of protonated hydroxylamine with cyanoacetaldehyde to give the most stable isomer of protonated cyanoacetamide, (O-prot) CNCH2COHNH2 +, from now on O-prot CAH+:
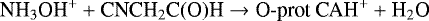 (3)
(3)
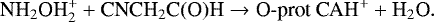 (4)
(4)
The energy profiles for both reactions were computed at B2PLYPD3 and CCSD(T) levels of theory, and they are depicted in Fig. 1. This figure shows that the results obtained with the B2PLYPD3 and CCSD(T) methods agree well.
For reaction of the most stable isomer of protonated hydroxylamine (NH3OH+), the process starts with the formation of the I1 intermediate, which is the result of the interaction between thecarbonylic oxygen of cyanoacetaldehyde with one of the hydrogen atoms of the NH3 group of protonated hydroxylamine. The exothermic formation of the I1 intermediate (ΔE = −18.0 kcal mol−1 at the CCSD(T) level) produces an energy reservoir that is used as the reaction proceeds toward the products. After I1 is formed, a hydrogen atom migrates from nitrogen to oxygen through the TS1 transition state, which is located 1.5 kcal mol−1 (755 K) above reactants (at the CCSD(T) level), giving the I2 intermediate. This intermediate evolves to the I3 intermediate through the TS2 transition state, which is located 25.3 kcal mol−1 (12 681 K), at the CCSD(T) level, above reactants. In the TS2 transition state, a water molecule is coordinated to the NH2 group. Thus, when the NH2 group is properly oriented, an N–C bond is formed, evolving to the formation of N-protonated cyanoacetamide and releasing a water molecule. Finally, an isomerization of the N-protonated form to the most stable isomer, O-prot CAH+, takes place through TS3, which is located − 13.9 kcal mol−1, at the CCSD(T) level, below reactants. In the TS3 transition state, a hydrogen atom of the NH3 group migrates to the carbonylic oxygen. The process can be summarized as follows:
![Mathematical equation: \begin{align*} &\textrm{NH}_3\textrm{OH}^+ + \textrm{CNCH}_2\textrm{CHO} \xrightarrow {\textrm{(TS1)}}\ \textrm{I1} \xrightarrow {\textrm{(TS2)}}\ \textrm{I2} \xrightarrow {\textrm{(TS3)}} \nonumber\\[4pt] &\quad{\textrm{N}}\hbox{-}\textrm{prot\ CAH}^+ \rightarrow {\textrm{O}}\hbox{-}\textrm{prot}\ \textrm{CAH}^+ + \textrm{H}_2\textrm{O}\ \textrm{(path\ 1)}.\nonumber \end{align*}](/articles/aa/full_html/2020/12/aa38766-20/aa38766-20-eq14.png)
As shown in Fig. 1, the global process is exothermic (ΔE = − 64.3 kcal mol−1 at the CCSD(T) level)), and it has a non-negligible activation barrier of 25.3 kcal mol−1 (12 732 K) at the CCSD(T) level of theory. This barrier precludes this mechanism from taking place in the ISM.
The first step in the mechanism starting from the less stable isomer of protonated hydroxylamine is the direct interaction between one of the hydrogen atoms bonded to oxygen of NH2OH2 + and the carbonylic oxygen of CNCH2C(O)H, giving the I2 intermediate. This complex, as in path 1, finally evolves through TS2 to produce N-protonated cyanoacetamide and H2 O, as is schematized in path 2. Finally, the N-protonated form can isomerize through the TS3 transition state to give CAH+ as the final product, which is located − 39.4 kcal mol−1, at the CCSD(T) level, below reactants:
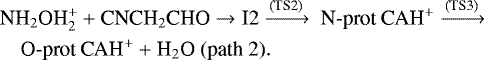
This reaction is a clear exothermic process (ΔE = −89.8 kcal mol−1 at the CCSD(T) level) without a net activation barrier because now the TS2 transition state lies 0.3 kcal mol−1 (at the CCSD(T) level) below reactants. Thus, the formation of CAH+ from the reaction of the less stable isomer of protonated hydroxylamine and cyanoacetaldehyde might be feasible under interstellar conditions. As we pointed out, although protonated COMs have not yet been detected in the ISM, it seems that they could evolve to neutral species by dissociative recombination. The neutral form, CH2(CN)C(O)NH2, might therefore be a candidate molecule to be searched for in the ISM. The optimized geometries at the B2PLYPD3/aug-cc-pVTZ level of theory for the intermediate species (I1 and I2) and transition states (TS1, TS2, and TS3) involved in the formation process of CAH+ are depictedin Fig. 2.
Relative energies (referred to reactants), including zero-point corrections, in kcal mol−1, obtained at different levels of theory for the reaction between protonated hydroxylamine and cyanoacetaldehyde yielding protonated cyanoacetamide.
 |
Fig. 1 Energy profile in kcal/mol for the reactions of protonated hydroxilamine with cyanoacetaldehyde producing O-protonated cyanoacetamide computed at the B2PLYPD3/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ (in parentheses) levels of theory. The zero-point vibrational energy computed at the B2PLYPD3/aug-cc-pVTZ level is included. |
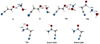 |
Fig. 2 Intermediates, transition states, and products involved on the reaction of formation of protonated cyanoacetamide. Geometries are optimized at the B3PLYPD3/aug-cc-pVTZ level of theory. Bond lengths are given in Angstroms for the intermediates and TS. |
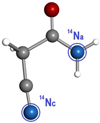 |
Fig. 3 Structure of trans cyanoacetamide, showing the two different 14N nuclei: an amide (Na) and a cyano (Nc) nitrogen atom. |
 |
Fig. 4 Rotational transitions 111 ← 000 and 212← 101 of trans cyanoacetamide observed during the LA-MB-FTMW experiment along with the B2PLYPD3/aug-cc-pVTZ predictions, displaying the high resolution of our spectrometer as well as the great agreement between the experimental and theoretically predicted frequencies. The resonance frequency is determined by the arithmetic mean of two Doppler components. The LA-MB-FTMW spectrum was obtained by averaging 200 experimental cycles (six free induction decay signals per cycle). |
Theoretical and experimental spectroscopic parameters for trans cyanoacetamide.
3.2 Microwave rotational spectrum of cyanoacetamide
After we found a feasible process to form protonated cyanoacetamide under interstellar conditions, we recorded the rotational spectrum of the neutral form, (CH2(CN)C(O)NH2), as a first step for its eventual radio-astronomical detection. The anticipated equilibrium structures, trans and gauche cyanoacetamide, are near prolate asymmetric tops. Recognizable patterns of μa -type R-branch transitions separated by approximately B + C frequency intervals are expected in the rotational spectrum. However, with the aid of high-level B2PLYPD3 computations, it was found that only the trans conformer was a real equilibrium structure onthe PES. A vibrational analysis for the gauche-conformer showed an imaginary frequency associated with the bending mode. We then focused on the analysis of trans cyanoacetamide (see Fig. 3). Cyanoacetamide is a challenging problem for high-resolution rotational studies. Its weak dipole moment (μa = 0.3 D and μb = 0.5 D) predicts very weak rotational transitions; additionally, the two nitrogen atoms divide the intensity of each rotational transition into several components due to the hyperfine structure. Therefore the experimental study of cyanoacetamide is difficult.
At the first stage of the line assignment, we scanned long frequency intervals of about 100 MHz around the predicted μa -type R-branch rotational transitions (with J′ ← J″ = 2 ← 1 and 3 ←2). Then, the polarization power was optimized according to the really low μa electric dipole-moment component (μa = 0.3 D). Finally, the 202 ← 101 and 303 ← 202 transitions were identified. Furthermore, all the observed rotational transitions display a complex hyperfine structure as a result of the two different 14N nuclei (see Fig. 4). This structure arises from the interaction of the electric quadrupole moment of the two 14N (I = 1) nuclei, with the electric field gradient created at the site of the quadrupolar nucleus by the remaining electronic and nuclear charges of the molecule. This interaction results in a coupling between the 14N nuclear spin and the overall angular momentum, which is shown as a characteristic hyperfine pattern in the rotational spectra (Gordy & Cook 1970). This coupling moreover leads to a splitting in the rotational energy levels that decreases the overall intensity of each rotational transition, which are already very weak because of the low dipole moment component. The initial assignment was then extended to other intense μb -type transitions. After iterative fitting and prediction, a total of 28 hyperfine components were collected and fit (Pickett et al. 1998) to a rigid rotor Hamiltonian, HR, supplemented with a term to take into account the quadrupole interaction HQ (Foley 1947; Robinson & Cornwell 1953), namely H = HR + HQ. The quadrupole coupling Hamiltonian was set up in the coupled basis set (I1, I2, I, J, K and F), where I1 + I2 = I, and I + J = F. The energy levels involved in each transition were thus labelled with the quantum numbers, Ka, Kc, I, and F. The resolution attained with our broadband LA-MB-FTMW technique was sufficient to fully resolve the hyperfine patterns. However, only the diagonal elements (χaa, χbb, and χcc) of the quadrupole coupling tensor χ were determined in the fit of the observed transitions within the estimated accuracy of the frequency measurements. The derived experimental and theoretical rotational and quadrupole coupling constants for trans cyanoacetamide are listed in Table 2, and the measured rotational transitions are collected in Table 3.
The first piece of evidence in the identification of the observed structure comes from the values of the rotational constants, which critically depend on the mass distribution of the molecule. Table 2 shows that the spectroscopic parameters predicted for the trans cyanoacetamide at the B2PLYPD37aug-cc-pVTZ level are very close to those experimentally determined. In this case, another interesting spectroscopic tool is taken into account: the two 14N nuclei in our molecule, which are also helpful to identify structures. In our predictions, we included the values of the quadrupole coupling constants for both 14Nc (cyano) and 14Na (amide) nuclei. It is known that the nuclear quadrupole-coupling interactions strongly depend on the electronic environment, position, and orientation of the 14N nuclei. The 14N nuclei introduce hyperfine rotational probes at defined sites of cyanoacetamide and act as a probe of the chemical environment of both Nc and Na quadrupolarnuclei. When we compare the experimental and theoretical rotational constants, factors ranging from 0.9999 to 1.0066 bring the values of predicted rotational constants of Table 2 to those experimentally obtained. Thus, we can infer that its actual geometry should be close to that calculated at the B2PLYPD3 level. The excellent match between the experimental and theoretical values shown in Table 2 enables the unambiguous identification of the observed form as trans cyanoacetamide. In conclusion, the microwave spectrum of the most stable form of cyanoacetamide has been studied up to 18 GHz to provide accurate laboratory reference spectra that can be used with confidence in future searches for cyanoacetamide in several low-frequency surveys.
Finally, we provide the rotational (Qr) and vibrational (Qv) partition functions of cyanoacetamide, which are shown in Table 4. We calculated the values of Qr from first principles at different temperatures, using the Picket program (Pickett et al. 1998). We estimated the vibrational part, Qv, using an anharmonic approximation and a simple formula that corresponds to Eq. (3.60) of Gordy & Cook (1970), where only the ten lowest vibrational modes were taken into account. We obtained the frequencies of the normal modes from double-hybrid calculations of both harmonic and anharmonic approximations, respectively, using the B2PLYPD3 method and the aug-cc-pVTZ basis set (see Table A.2). The full partition function, Qtot, is therefore the product of Qr and Qv. Finally, the complete list of transitions of the calculated reference spectra is available at the CDS.
Complete list of the measured transition frequencies for the ground-state of cyanoacetamide.
Rotational and vibrational partition functions of cyanoacetamide.
4 Conclusions
We have carried out a theoretical study of the formation process of protonated cyanoacetamide as a plausible cyanoacetamide precursor, obtained from reaction between protonated hydroxylamine and cyanoacetaldehyde. A detailed analysis of the corresponding singlet potential energy surface has been performed. Thus, we found an exothermic process (ΔE = − 89.8 kcal mol−1 at the CCSD(T) level) with no net activation barrier initiated by the high-energy isomer of protonated hydroxylamine, which leads to protonated cyanoacetamide. Our results suggest that the neutral form, (CH2(CN)C(O)NH2), might be a candidate molecule to be searched for in the ISM.
Furthermore, we presented the first high-resolution rotational characterization of trans cyanoacetamide. The capability provided by the laser ablation coupled with time domain Fourier transform microwave techniques to obtain very accurate spectroscopic constants in combinationwith high-level theoretical computations enables the unequivocal identification of this elusive molecule. The two 14Nc and 14Na nuclei of cyanoacetamide act as hyperfine rotational probes of molecular conformation that increase the usefulness of this spectroscopic technique even more. The experimental rotational constants together with the values of the 14N nuclear quadrupole coupling constants presented in this work are provided in order to reproduce the spectrum perfectly, and they constitute a first step to identify this molecule in the ISM. We hope this work will benefit the astrophysics community by allowing a possible detection in the near future of cyanoacetamide, whose rotational signatures have remained unknown until now.
By the time of the submission of the paper, Rivilla et al. (2020) has just reported the first detection of hydroxylamine (NH2OH) in the ISM toward the quiescent molecular cloud G+0.693-0.027 located in the Galactic Center.
Acknowledgements
This research was supported by the Ministerio de Ciencia e Innovación (CTQ2016-76393-P), the Ministerio de Economia Industria y Competitividad (Grant AYA2017-87515-P) Junta de Castilla y León (Grants VA077U16 and Grant VA010G18), and the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC-2013-SyG, Grant Agreement no. 610 256 NANOCOSMOS, is gratefully acknowledged. M.S.N. acknowledges funding from the Spanish “Ministerio de Ciencia, Innovación y Universidades” under predoctoral FPU Grant (FPU17/02987).
Appendix A Complementary tables
Table A.1 provides the centrifugal distortion parameters and Table A.2 lists the theoretical frequencies of the lowest vibrational modes.
Quartic and sextic centrifugal distortion parameters for trans cyanoacetamide (S-reduction, Ir -representation).
Calculated harmonic and anharmonic frequencies together with the corresponding infrared intensities of the lowest vibrational modes.
Appendix B Complementary reactions and figures
Figure B.1 shows the energy profile corresponding to the reaction between vinyl cyanide, CH2CHCN (VC) and O(3P) radical to give cyanoacetaldehyde. This is an exothermic associative reaction that requires an intersystem spin crossing from the triplet state of the VC-O intermediate to the final single state of cyanoacetaldehyde. The reaction starts by forming a stable compound, 3 [VC-O], between the vinylcyanide molecule and the oxygen atom, in its triplet state. The formation of the triplet intermediate leads to a stabilization of 27.3 kcal mol−1 (at the CCSD(T)/aug-cc-pVTZ level). 3[VC-O] is a triplet intermediate from which two products, in their singlet state, can be generated by a spin crossing: (1) cyanoacetaldehyde, which can be formed by simple hydrogen migration, and (2) oxirane-2-carbonitrile, formed by aclosure of the O–C–C 3-member ring. To obtain these two products, a spin transition is required. The change in spin multiplicity leads to a new intermediate, which is a singlet-state diradical 1 [VC-O]. Depending on the value of the OCCH dihedral angle, this intermediate will finally evolve to oxirane-2-carbonitrile (CH2OCHCN) or to cyanoacetaldehyde (CHOCH2CN), which is the most stable product, and it is located 102.2 kcal mol−1 below reactants. From a purely thermodynamical point of view, cyanoacetaldehyde should be obtained. That the reaction needs an intersystem crossing to occur means that the reaction might be a slowly occurring but also a plausible process under astrophysical conditions. Furthermore, the singlet complex 1 [VC-O] would require a further energetic stabilization by photon emission or even from the environment. A similar process was studied in the reaction of propylene and O (3P), which led to propanol instead of the targeted propylene oxide (Bodo et al. 2019).
 |
Fig. B.1 Energy profile in kcal/mol for the reactions of vinyl cyanide with O (3P) radical producing cyanoacetaldehyde computed at the B2PLYPD3/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ (in parentheses) levels of theory. The zero-point vibrational energy computed at the B2PLYPD3/aug-cc-pVTZ level is included. |
References
- Agúndez, M., Marcelino, N., & Cernicharo, J. 2018, ApJ, 861, L22 [NASA ADS] [CrossRef] [Google Scholar]
- Alonso, J. L., & López, J. C. 2015, in Gas-Phase IR Spectroscopy and Structure of Biological Molecules (Cham: Springer International Publishing), 335 [Google Scholar]
- Alonso, J. L., Pérez, C., Eugenia Sanz, M., et al. 2009, Phys. Chem. Chem. Phys., 11, 617 [CrossRef] [Google Scholar]
- Alonso, J. L., Lozoya, M. A. I., Peña, López, J. C., et al. 2014, Chem. Sci., 5, 515 [CrossRef] [Google Scholar]
- Alonso, E. R., Kolesniková, L., & Alonso, J. L. 2017, J. Chem. Phys., 147, 124312 [CrossRef] [Google Scholar]
- Alonso, E. R., Kolesniková, L., Białkowska-Jaworska, E., et al. 2018, ApJ, 861, 70 [CrossRef] [Google Scholar]
- Barrientos, C., Redondo, P., Largo, L., et al. 2012, ApJ, 748, 99 [NASA ADS] [CrossRef] [Google Scholar]
- Becke, A. D. 1993, J. Chem. Phys., 98, 5648 [NASA ADS] [CrossRef] [Google Scholar]
- Bell, M. B., Feldman, P. A., Travers, M. J., et al. 1997, ApJ, 483, L61 [NASA ADS] [CrossRef] [Google Scholar]
- Belloche, A., Menten, K. M., Comito, C., et al. 2008, A&A, 482, 179. [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Belloche, A., Garrod, R. T., Müller, H. S. P., et al. 2014, Science, 345, 1584 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Belloche, A., Meshcheryakov, A. A., Garrod, R. T., et al. 2017, A&A, 601, A49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bermudez, C., Mata, S., Cabezas, C., Alonso, J. L. 2014, Angew. Chem. Int. Ed. Engl., 53, 11015 [CrossRef] [Google Scholar]
- Bockelée-Morvan, D., Lis, D. C., Wink, J. E., et al. 2000, A&A, 353, 1101 [Google Scholar]
- Bodo, E., Bovolenta, G., Simha, C., Spezia, R., 2019, ThChA, 138, 97 [Google Scholar]
- Bogey, M., Dubus, H., & Guillemin, J. C. 1990, J. Molec. Spectr., 143, 180 [NASA ADS] [CrossRef] [Google Scholar]
- Broten, N. W., Oka, T., Avery, L. W., et al. 1978, ApJ, 223, L105 [NASA ADS] [CrossRef] [Google Scholar]
- Cabezas, C., Varela, M., Peña, I., et al. 2012, Phys. Chem. Chem. Phys., 14, 13618 [CrossRef] [Google Scholar]
- Congiu, E., Fedoseev, G., Ioppolo, S., et al. 2012, ApJ, 750, L12 [NASA ADS] [CrossRef] [Google Scholar]
- Coustenis, A., Bezard, B., Gautier, D., et al. 1991, Icarus, 89, 152 [NASA ADS] [CrossRef] [Google Scholar]
- Coustenis, A., Schmitt, B., Khanna, R. K., et al. 1999, Planet. Space Sci., 47, 1305 [NASA ADS] [CrossRef] [Google Scholar]
- Dunning, T. H. 1989, J. Chem. Phys., 90, 1007 [NASA ADS] [CrossRef] [Google Scholar]
- El Bialy, S. A. A., & Gouda, M.A. 2011, J. Heterocyclic Chem., 48, 1280 [CrossRef] [Google Scholar]
- Ferris, J. P., Sánchez, R. A., Orgel, L. E. 2006, J. Mol. Biol., 33, 693 [CrossRef] [Google Scholar]
- Foley, H. M. 1947, Phys. Rev., 71, 747 [CrossRef] [Google Scholar]
- Gaussian 16, Revision A.03, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J., 2016 Gaussian, Inc., Wallingford CT [Google Scholar]
- Godfrey, P. D., Brown, R. D., Robinson, B. J., et al. 1973, Astrophys. Lett., 13, 119 [Google Scholar]
- Gonzalez, C., & Schlegel, H. B. 1991, J. Chem. Phys., 95, 5853 [CrossRef] [Google Scholar]
- Gordy, W., & Cook, R. L. 1970, Microwave Molecular Spectra (New York: Interscience Pub.) [Google Scholar]
- Grimme, S., Antony, J., Ehrlich, S., et al. 2010, J. Chem. Phys., 132, 154104 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- He, J., Vidali, G., Lemaire, J.-L., et al. 2015, ApJ, 799, 49 [NASA ADS] [CrossRef] [Google Scholar]
- Herbst, E., & van Dishoeck, E. F. 2009, ARA&A, 47, 427 [NASA ADS] [CrossRef] [Google Scholar]
- Hollis, J. M., Lovas, F. J., Remijan, A. J., et al. 2006, ApJ, 643, L25 [Google Scholar]
- Horn, A., Møllendal, H., & Guillemin, J.-C. 2008, J. Phys. Chem. A, 112, 11009 [CrossRef] [Google Scholar]
- Hoy, A. R., Mills, I. M., & Strey, G. 1972, Mol. Phys., 24, 1265 [NASA ADS] [CrossRef] [Google Scholar]
- Kawaguchi, K., Kasai, Y., Ishikawa, S.-I., et al. 1994, ApJ, 420, L95 [NASA ADS] [CrossRef] [Google Scholar]
- Kolesniková, L., Alonso, E. R., Mata, S., et al. 2017a, ApJS, 229, 26 [CrossRef] [Google Scholar]
- Kolesniková, L., Alonso, E. R., Mata, S., et al. 2017b, ApJS, 233, 24 [NASA ADS] [CrossRef] [Google Scholar]
- Kołos, R., & Grabowski, Z. R. 2000, Ap&SS, 271, 65 [NASA ADS] [CrossRef] [Google Scholar]
- Kunde, V. G., Aikin, A. C., Hanel, R. A., et al. 1981, Nature, 292, 686 [NASA ADS] [CrossRef] [Google Scholar]
- Largo, L., Rayón, V. M., Barrientos, C., et al. 2009, Chem. Phys. Lett., 476, 174 [CrossRef] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- León, I., Alonso, E. R., Mata, S., et al. 2017, Phys. Chem. Chem. Phys., 19, 24985 [CrossRef] [Google Scholar]
- Lovas, F. J., Remijan, A. J., Hollis, J. M., et al. 2006, ApJ, 637, L37 [NASA ADS] [CrossRef] [Google Scholar]
- Møllendal, H., Margulès, L., Motiyenko, R. A., et al. 2012, J. Phys. Chem. A, 116, 4047 [CrossRef] [Google Scholar]
- Møller, C., & Plesset, M. S. 1934, Phys. Rev., 46, 618. [NASA ADS] [CrossRef] [Google Scholar]
- Nishi, N., Shinohara, H., & Okuyama, T. 1984, J. Chem. Phys., 80, 3898 [NASA ADS] [CrossRef] [Google Scholar]
- Pardo, J. R., Cernicharo, J., Goicoechea, J. R., et al. 2005, Astrochem.: Rec. Successes Curr. Challenges, 40 [Google Scholar]
- Petrie, S., Millar, T. J., & Markwick, A. J. 2003, MNRAS, 341, 609 [NASA ADS] [CrossRef] [Google Scholar]
- Pickett, H. M., Poynter, R. L., Cohen, E. A., et al. 1998, J. Quant. Spec. Rad. Transf., 60, 883 [Google Scholar]
- Raghavachari, K., Trucks, G. W., Pople, J. A., et al. 1989, Chem. Phys. Lett., 157, 479 [NASA ADS] [CrossRef] [Google Scholar]
- Redondo, P., Barrientos, C., & Largo, A. 2014, ApJ, 793, 32 [NASA ADS] [CrossRef] [Google Scholar]
- Redondo, P., Largo, A., & Barrientos, C. 2015, A&A, 579, A125 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Redondo, P., Martínez, H., Largo, A., et al. 2017, A&A, 603, A139 [CrossRef] [EDP Sciences] [Google Scholar]
- Rivilla, V. M., Martín-Pintado, J., Jiménez-Serra, I., et al. 2020, ApJ, 899, L28 [CrossRef] [Google Scholar]
- Robinson, G. W., & Cornwell, C. D. 1953, J. Chem. Phys., 21, 1436 [NASA ADS] [CrossRef] [Google Scholar]
- Sánchez, R. A., Ferris, J. P., Orgel, L. E. 1967, J. Mol. Biol. 33, 693 [Google Scholar]
- Sanz-Novo, M., Belloche, A., Alonso, J. L., et al. 2020, A&A, 639, A135 [CrossRef] [EDP Sciences] [Google Scholar]
- Schwabe, T., & Grimme, S. 2007, Phys. Chem. Chem. Phys., 9, 3397 [CrossRef] [PubMed] [Google Scholar]
- Schermann, J.-P. 2008 Spectroscopy and Modeling of Biomolecular Building Blocks (Amsterdam: Elsevier) [Google Scholar]
- Snow, J. L., Orlova, G., Blagojevic, V., Bohme, D. K. 2007, J. Am. Chem. Soc., 129, 9910 [CrossRef] [PubMed] [Google Scholar]
- Tsegaw, Y. A., Góbi, S., Förstel, M., et al. 2017, J. Phys. Chem. A, 121, 7477 [CrossRef] [Google Scholar]
- van Haverbeke, L., & Herman, M. A. 1975, Spectrochim. Acta Part A, 31, 959 [CrossRef] [Google Scholar]
- Winnewisser, G., & Walmsley, C. M. 1978, A&A, 70, L37 [NASA ADS] [Google Scholar]
- Woon, D. E., & Dunning, T. H. 1993, J. Chem. Phys., 98, 1358 [CrossRef] [Google Scholar]
- Zheng, W., & Kaiser, R. I. 2010, J. Phys. Chem. A, 114, 5251 [CrossRef] [Google Scholar]
All Tables
Relative energies (referred to reactants), including zero-point corrections, in kcal mol−1, obtained at different levels of theory for the reaction between protonated hydroxylamine and cyanoacetaldehyde yielding protonated cyanoacetamide.
Complete list of the measured transition frequencies for the ground-state of cyanoacetamide.
Quartic and sextic centrifugal distortion parameters for trans cyanoacetamide (S-reduction, Ir -representation).
Calculated harmonic and anharmonic frequencies together with the corresponding infrared intensities of the lowest vibrational modes.
All Figures
|
Fig. 1 Energy profile in kcal/mol for the reactions of protonated hydroxilamine with cyanoacetaldehyde producing O-protonated cyanoacetamide computed at the B2PLYPD3/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ (in parentheses) levels of theory. The zero-point vibrational energy computed at the B2PLYPD3/aug-cc-pVTZ level is included. |
| In the text | |
|
Fig. 2 Intermediates, transition states, and products involved on the reaction of formation of protonated cyanoacetamide. Geometries are optimized at the B3PLYPD3/aug-cc-pVTZ level of theory. Bond lengths are given in Angstroms for the intermediates and TS. |
| In the text | |
|
Fig. 3 Structure of trans cyanoacetamide, showing the two different 14N nuclei: an amide (Na) and a cyano (Nc) nitrogen atom. |
| In the text | |
|
Fig. 4 Rotational transitions 111 ← 000 and 212← 101 of trans cyanoacetamide observed during the LA-MB-FTMW experiment along with the B2PLYPD3/aug-cc-pVTZ predictions, displaying the high resolution of our spectrometer as well as the great agreement between the experimental and theoretically predicted frequencies. The resonance frequency is determined by the arithmetic mean of two Doppler components. The LA-MB-FTMW spectrum was obtained by averaging 200 experimental cycles (six free induction decay signals per cycle). |
| In the text | |
|
Fig. B.1 Energy profile in kcal/mol for the reactions of vinyl cyanide with O (3P) radical producing cyanoacetaldehyde computed at the B2PLYPD3/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ (in parentheses) levels of theory. The zero-point vibrational energy computed at the B2PLYPD3/aug-cc-pVTZ level is included. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.