| Issue |
A&A
Volume 601, May 2017
|
|
|---|---|---|
| Article Number | A49 | |
| Number of page(s) | 41 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201629724 | |
| Published online | 27 April 2017 | |
Rotational spectroscopy, tentative interstellar detection, and chemical modeling of N-methylformamide⋆
1 Max-Planck-Institut für Radioastronomie, Auf dem Hügel 69, 53121 Bonn, Germany
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2 Microwave spectroscopy department, Institute of Radio Astronomy of NASU, Chervonopraporna Str. 4, 61002 Kharkiv, Ukraine
3 Departments of Chemistry and Astronomy, University of Virginia, Charlottesville, VA 22904, USA
4 Quantum Radiophysics Department of V. N. Karazin Kharkiv National University, Svobody Square 4, 61022 Kharkiv, Ukraine
5 Laboratoire de Physique des Lasers, Atomes, et Molécules, UMR 8523, CNRS, Université de Lille 1, 59655 Villeneuve d’Ascq Cedex, France
6 I. Physikalisches Institut, Universität zu Köln, Zülpicher Str. 77, 50937 Köln, Germany
Received: 16 September 2016
Accepted: 17 January 2017
Abstract
Context. N-methylformamide, CH3NHCHO, may be an important molecule for interstellar pre-biotic chemistry because it contains a peptide bond, which in terrestrial chemistry is responsible for linking amino acids in proteins. The rotational spectrum of the most stable trans conformer of N-methylformamide is complicated by strong torsion-rotation interaction due to the low barrier of the methyl torsion. For this reason, the theoretical description of the rotational spectrum of the trans conformer has, up to now, not been accurate enough to provide a firm basis for its interstellar detection.
Aims. In this context, as a prerequisite for a successful interstellar detection, our goal is to improve the characterization of the rotational spectrum of N-methylformamide.
Methods. We use two absorption spectrometers in Kharkiv and Lille to measure the rotational spectra over the frequency range 45–630 GHz. The analysis is carried out using the Rho-axis method and the RAM36 code. We search for N-methylformamide toward the hot molecular core Sagittarius (Sgr) B2(N2) using a spectral line survey carried out with the Atacama Large Millimeter/submillimeter Array (ALMA). The astronomical spectra are analyzed under the assumption of local thermodynamic equilibrium. The astronomical results are put into a broader astrochemical context with the help of a gas-grain chemical kinetics model.
Results. The new laboratory data set for the trans conformer of N-methylformamide consists of 9469 distinct line frequencies with J ≤ 62, including the first assignment of the rotational spectra of the first and second excited torsional states. All these lines are fitted within experimental accuracy for the first time. Based on the reliable frequency predictions obtained in this study, we report the tentative detection of N-methylformamide toward Sgr B2(N2). We find N-methylformamide to be more than one order of magnitude less abundant than formamide (NH2CHO), a factor of two less abundant than the unsaturated molecule methyl isocyanate (CH3NCO), but only slightly less abundant than acetamide (CH3CONH2). We also report the tentative detection of the 15N isotopolog of formamide (15NH2CHO) toward Sgr B2(N2). The chemical models indicate that the efficient formation of HNCO via NH + CO on grains is a necessary step in the achievement of the observed gas-phase abundance of CH3NCO. Production of CH3NHCHO may plausibly occur on grains either through the direct addition of functional-group radicals or through the hydrogenation of CH3NCO.
Conclusions. Provided the detection of N-methylformamide is confirmed, the only slight underabundance of this molecule compared to its more stable structural isomer acetamide and the sensitivity of the model abundances to the chemical kinetics parameters suggest that the formation of these two molecules is controlled by kinetics rather than thermal equilibrium.
Key words: line: identification / molecular data / radio lines: ISM / ISM: molecules / ISM: individual objects: Sagittarius B2(N) / astrochemistry
Full Tables 2 and 3 are only available at the CDS via anonymous ftp to cdsarc.u-strasbg.fr (130.79.128.5) or via http://cdsarc.u-strasbg.fr/viz-bin/qcat?J/A+A/601/A49
© ESO, 2017
1. Introduction
The peptide bond is a fundamental building block of life on Earth (Kaiser et al. 2013). Therefore, peptide molecules have for a long time attracted much attention. The simplest molecule containing a peptide bond, formamide (NH2CHO), was detected in the interstellar medium (ISM) back in the 1970s (Rubin et al. 1971). The relatively high abundance of formamide also permitted the detection of rotational lines of its first excited vibrational state ν12 = 1 in Orion KL (Motiyenko et al. 2012) and in Sagittarius (Sgr) B2(N) (Belloche et al. 2013). N-methylformamide, CH3NHCHO, is one of the simplest derivatives of formamide and also a peptide molecule. It is of interest as a candidate for interstellar detection because its structural isomer acetamide (CH3CONH2) has already been detected in the ISM (Hollis et al. 2006; Halfen et al. 2011). CH3NHCHO is the second most stable C2H5NO isomer after acetamide (Lattelais et al. 2010).
N-methylformamide exists in two stable rotameric forms, trans and cis. Their structures are shown in Fig. 1. According to quantum chemical calculations, the trans conformer is more stable than cis by 466 cm-1 (666 K, Kawashima et al. 2010). The trans conformer is also characterized by a very low barrier to internal rotation of the methyl top. The coupling between the overall rotation of the molecule and the almost free rotation of the methyl top significantly complicates the description of the spectrum. For this reason, N-methylformamide has been the subject of extensive spectroscopic investigations, but for a long time the analysis of its microwave rotational spectrum did not yield satisfactory results. Fantoni & Caminati (1996) were the first to succeed in assigning rotational spectral lines belonging to the trans conformer in the CH3 internal rotation ground state of A symmetry. They performed measurements between 18 and 40 GHz, but could not identify any spectral lines of the E species. Later, Fantoni et al. (2002) published results of measurements and analysis for E lines. The V3 barrier of the methyl group internal rotation was determined to be 55.17 ± 0.84 cm-1.
Recently, Kawashima et al. (2010) carried out a new spectroscopic investigation of N-methylformamide. In that study, rotational spectra of both trans and cis conformers of the normal as well as deuterated CH3NHCHO isotopologs were measured in the frequency range of 5–118 GHz. Molecular parameters including rotational constants and V3 barriers to methyl-group internal rotation were determined for all investigated species and conformers. Owing to the relatively high barrier to internal rotation for the cis conformer, a good description was obtained for its 108 measured transitions. However, the low barrier and the limitations in the model used for the theoretical description of the rotational spectrum allowed these authors to fit only low J quantum number transitions (J < 11) for the trans conformer. In addition, 60 out of 467 assigned transitions of the trans conformer were excluded from the final fit as their residuals ranged from 1 to 67 MHz and were much higher than the experimental accuracy estimated to be 0.004–0.05 MHz.
One should note that in previous publications, two different schemes were used for naming the conformations of N-methylformamide. Fantoni & Caminati (1996) and Fantoni et al. (2002) used the dihedral angle D(H−N−C−H′) where H′ is the carbonyl group hydrogen. The conformer with D = 0° was named cis and the conformer with D = 180° was named trans. Kawashima et al. (2010) used another convention widely accepted for molecules with a peptide bond. According to this convention, applied for N-methylformamide, one should use the D∗(Y−N−C−X) dihedral angle, where X is the carbonyl hydrogen and Y is the methyl group. Cis and trans conformers named using this convention are thus the opposite of the cis and trans conformers in Fantoni & Caminati (1996) and Fantoni et al. (2002). Here we use the naming adopted by Kawashima et al. (2010), that is, using the D∗ dihedral angle.
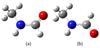 |
Fig. 1 Structure of trans (a) and cis (b) conformations of N-methylformamide. |
In the present study, we extend the measurement and analysis of the rotational spectrum of trans N-methylformamide, which is of higher interest for astrophysical detection. We use the accurate frequency predictions obtained in this study to search for N-methylformamide in the ISM. We target the high mass star-forming region Sgr B2(N), one of the most prolific sources for the detection of complex organic molecules in the ISM (e.g., Belloche et al. 2013). For this, we use a spectral survey of Sgr B2(N) conducted with the Atacama Large Millimeter/submillimeter Array (ALMA) in its Cycles 0 and 1. This survey aims at exploring molecular complexity with ALMA (EMoCA, see Belloche et al. 2016).
The experimental setup is presented in Sect. 2. The analysis of the rotational spectrum of N-methylformamide and the results that follow are described in Sect. 3. A tentative detection of N-methylformamide in Sgr B2(N) is presented in Sect. 4 along with the derivation of column densities of other related molecules of interest. Chemical modeling is performed in Sect. 5 to put the observational results into a broader astrochemical context. The results are discussed in Sect. 6 and the conclusions are presented in Sect. 7.
2. Experimental setup
A sample of N-methylformamide was purchased from Sigma-Aldrich and used without further purification. The experimental investigation of the absorption spectrum of N-methylformamide was carried out over the frequency range 45–630 GHz using two microwave spectrometers. The first one is the automated millimeter wave spectrometer of the Institute of Radio Astronomy of NASU in Kharkiv, Ukraine (Alekseev et al. 2012). The second one is the terahertz spectrometer of the Laboratory of Physics of Lasers, Atoms, and Molecules in Lille, France (Zakharenko et al. 2015).
The spectrometer in Kharkiv is built according to the classical scheme of absorption spectrometers, and its detailed description can be found in Alekseev et al. (2012). The spectrometer was slightly upgraded with the aim of expanding the operating frequency range; a new backward-wave oscillator (BWO) unit covering frequencies from 34 to 52 GHz has been put into operation. Thus, at present, this spectrometer can record spectra between 34 and 250 GHz. In order to improve the sensitivity below 50 GHz, a new waveguide absorbing cell (a copper waveguide of 10 × 72 mm2 internal cross section and 295 cm length) was used instead of the commonly employed quasi-optic absorption cell. The measurements of N-methylformamide with the Kharkiv spectrometer were done in the frequency range 45 to 150 GHz. All measurements were performed at room temperature and with sample pressures (approximately 10 mTorr) that provided close to Doppler-limited spectral resolution. The frequency determination errors were estimated to be 10, 30, and 100 kHz depending on the measured signal-to-noise ratio.
The measurements in Lille were performed between 150 and 630 GHz at typical pressures of 10 Pa and at room temperature. The frequency determination errors were estimated to be 30 kHz and 50 kHz below and above 330 GHz, respectively. The frequencies of the lines with poor signal-to-noise ratio or distorted lineshape were measured with 50 kHz or 100 kHz accuracy.
3. Spectroscopic analysis and results
We performed the analysis using the Rho-axis method (RAM), which was already applied successfully to a number of molecules with large-amplitude torsional motion. The method uses the axis system obtained by rotation of the principal axis system to make the new z axis parallel to the ρ vector. The coordinates of the ρ vector are calculated using the following expression:
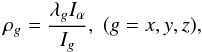 where λg are the direction cosines of the internal rotation axis of the top in the principal axis system, Ig are the principal inertia moments, and Iα is the inertia moment of the methyl top. The RAM Hamiltonian may be written as (Kleiner 2010):
where λg are the direction cosines of the internal rotation axis of the top in the principal axis system, Ig are the principal inertia moments, and Iα is the inertia moment of the methyl top. The RAM Hamiltonian may be written as (Kleiner 2010): 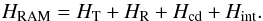 (1)HT represents the torsional Hamiltonian defined as:
(1)HT represents the torsional Hamiltonian defined as: 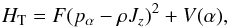 (2)where F is the internal rotation constant, pα is the internal rotation angular momentum conjugate to the torsion angle α, and V(α) is the internal rotation potential function:
(2)where F is the internal rotation constant, pα is the internal rotation angular momentum conjugate to the torsion angle α, and V(α) is the internal rotation potential function: 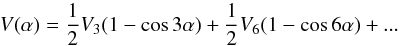 (3)HR represents the rigid rotor rotational Hamiltonian in the rho-axis system. In addition to usual A, B, and C terms, for the molecules with (xz) plane of symmetry (as is appropriate for N-methylformamide), it contains a non-diagonal term Dxz. In the Ir coordinate representation, HR has the following form:
(3)HR represents the rigid rotor rotational Hamiltonian in the rho-axis system. In addition to usual A, B, and C terms, for the molecules with (xz) plane of symmetry (as is appropriate for N-methylformamide), it contains a non-diagonal term Dxz. In the Ir coordinate representation, HR has the following form: 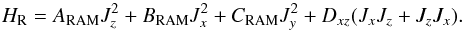 (4)The last two terms in Eq. (1), Hcd and Hint, are the usual centrifugal distortion and higher-order torsional-rotational interaction Hamiltonians, respectively.
(4)The last two terms in Eq. (1), Hcd and Hint, are the usual centrifugal distortion and higher-order torsional-rotational interaction Hamiltonians, respectively.
To fit and predict the rotational spectra, we used the RAM36 (Rho-axis method for 3 and 6-fold barriers) program that allows one to include in the model almost any symmetry-allowed torsion-rotation Hamiltonian term up to the twelfth order (Ilyushin et al. 2010, 2013). The RAM Hamiltonian in Eq. (1) may be expressed in the following form used in the RAM36 program : ![Mathematical equation: \begin{eqnarray} \label{ram36} \begin{split} H = & \frac{1}{2} \sum \limits_{knpqrs} B_{knpqrs0}\left[J^{2k} J_z^n J_x^p J_y^q p_\alpha^r \cos (3s\alpha) \right.\\ & \left. + \cos (3s\alpha)p_\alpha^r J_y^q J_x^p J_z^n J^{2k} \right] \\ & + \frac{1}{2} \sum \limits_{knpqrt} B_{knpqr0t}\left[J^{2k} J_z^n J_x^p J_y^q p_\alpha^r \sin (3t\alpha) \right.\\ & \left. + \sin (3t\alpha)p_\alpha^r J_y^q J_x^p J_z^n J^{2k} \right], \end{split} \end{eqnarray}](/articles/aa/full_html/2017/05/aa29724-16/aa29724-16-eq45.png) (5)where the Bknpqrst are fitting parameters. In the case of a C3v top and Cs frame (as is appropriate for N-methylformamide), the allowed terms in the torsion-rotation Hamiltonian must be totally symmetric in the group G6 (and also must be Hermitian and invariant to the time reversal operation). Since all individual operators pα, Jx, Jy, Jz, J2, cos(3sα), and sin(3tα) used in Eq. (5) are Hermitian, all possible terms provided by Eq. (5) will automatically be Hermitian. The particular term to be fitted is represented in the input file with a set of k,n,p,q,r,s,t integer indices that are checked by the program for conformity with time reversal and symmetry requirements, to prevent accidental introduction of symmetry-forbidden terms into the Hamiltonian. For example, B0000200 corresponds to F in Eq. (2), B0200000 to ARAM in Eq. (4), etc. In Table 1, which presents the final set of molecular parameters, we give, instead of Bknpqrst, more conventional names for the parameters whose nomenclature is based on the subscript procedures of Xu et al. (2008).
(5)where the Bknpqrst are fitting parameters. In the case of a C3v top and Cs frame (as is appropriate for N-methylformamide), the allowed terms in the torsion-rotation Hamiltonian must be totally symmetric in the group G6 (and also must be Hermitian and invariant to the time reversal operation). Since all individual operators pα, Jx, Jy, Jz, J2, cos(3sα), and sin(3tα) used in Eq. (5) are Hermitian, all possible terms provided by Eq. (5) will automatically be Hermitian. The particular term to be fitted is represented in the input file with a set of k,n,p,q,r,s,t integer indices that are checked by the program for conformity with time reversal and symmetry requirements, to prevent accidental introduction of symmetry-forbidden terms into the Hamiltonian. For example, B0000200 corresponds to F in Eq. (2), B0200000 to ARAM in Eq. (4), etc. In Table 1, which presents the final set of molecular parameters, we give, instead of Bknpqrst, more conventional names for the parameters whose nomenclature is based on the subscript procedures of Xu et al. (2008).
The RAM36 program uses the free-rotor quantum number m to label the torsional energy levels. It is well known that the internal rotation of a methyl top attached to a molecular frame should be treated as an anharmonic vibrational motion well below the top of the barrier to internal rotation, and as a nearly free internal rotation motion well above the top of the barrier. The vt labeling assumes that the spacings between degenerate and non-degenerate levels of the torsional Hamiltonian associated with a given vt are much smaller than those between levels with different vt (Lin & Swalen 1959). The trans conformation of N-methylformamide represents an intermediate case because of the relatively low barrier to internal rotation (Lin & Swalen 1959). Owing to the low barrier, the A−E splitting in the first excited torsional state vt = 1 of the trans conformation is comparable with the energy difference between the vt = 0 and vt = 1 states, whereas the vt = 2 state lies well above the barrier. Because of this intermediate situation we decided to keep the quantum number labeling of torsional states m in Tables 2 and 3, which is used inside the RAM36 program.
The RAM36 code was modified to take into account the quadrupole hyperfine structure of the transitions that is present in the spectrum of N-methylformamide due to the non-zero electric quadrupole moment of the nitrogen atom. We used the standard hyperfine energy expression: ![Mathematical equation: \begin{eqnarray} \label{ehf} \begin{split} E_{hf} & = \left[ \chi_{aa}\left\langle J_a^2\right\rangle + \chi_{bb}\left\langle J_b^2\right\rangle - \left( \chi_{aa}+\chi_{bb} \right) \left\langle J_c^2\right\rangle \right. \\ & \left. \vphantom{J_c^2} + \chi_{ab}\left\langle \vphantom{J_c^2} J_aJ_b+J_bJ_a\right\rangle \right] \frac{2f(I,J,F)}{J(J+1)}, \end{split} \end{eqnarray}](/articles/aa/full_html/2017/05/aa29724-16/aa29724-16-eq66.png) (6)where f(I,J,F) is the Casimir function. Typically, a resolved pattern of the hyperfine structure was observed as a doublet with an approximately two-to-one ratio in intensities. The stronger doublet component contains unresolved hyperfine transitions with selection rules F = J + 1 ← F = J + 1 and F = J−1 ← F = J−1, whereas the weaker doublet component corresponds to the F = J ← F = J transition.
(6)where f(I,J,F) is the Casimir function. Typically, a resolved pattern of the hyperfine structure was observed as a doublet with an approximately two-to-one ratio in intensities. The stronger doublet component contains unresolved hyperfine transitions with selection rules F = J + 1 ← F = J + 1 and F = J−1 ← F = J−1, whereas the weaker doublet component corresponds to the F = J ← F = J transition.
We started our analysis of the trans N-methylformamide spectrum from refitting the data available from the literature (Fantoni & Caminati 1996; Fantoni et al. 2002; Kawashima et al. 2010) using the RAM36 code. Application of the RAM36 code allowed us to fit, almost within experimental error, the data available in the literature including those lines that were previously excluded from the fits due to relatively high observed-minus-calculated values (Kawashima et al. 2010). Thus we obtained a reliable basis for assigning the newly measured lines in the 45–630 GHz range. Assigning and fitting of the new data using the RAM36 program proceeded in a fairly conventional, iterative way going up in frequency.
Molecular parameters of the trans conformer of N-methylformamide obtained with the RAM36 program.
The complete data set treated at the final stage of the current study includes both our new data and data from the literature (Fantoni & Caminati 1996; Fantoni et al. 2002; Kawashima et al. 2010). The data set contains 12 456 A- and E-type transitions with J ≤ 62 and Ka ≤ 21 for trans N-methylformamide in the lowest three torsional states. Due to blending, these 12 456 transitions correspond to 9469 distinct line frequencies (mainly due to non-fully resolved quadrupole hyperfine structure). The fit chosen as the “best” uses a model consisting of 103 parameters. The weighted root-mean-square deviation of the fit of 12 456 microwave transition frequencies with J ≤ 62 is 0.84, indicating that assumed statistical uncertainties were slightly overestimated. The largest residual of 0.293 MHz is observed in the fit for the vt = 0E-symmetry species transition 499,41 ← 489,40. The final set of molecular parameters is presented in Table 1. The final data set of fitted transitions of the N-methylformamide trans conformer is presented in Table 2, where we provide quantum numbers for each level, followed by observed transition frequencies, measurement uncertainties, and residuals from the fit. The complete version of Table 2 is available at the CDS; here only part of the table is presented for illustration purposes.
Measured transitions of trans N-methylformamide in the vt = 0, 1, and 2 states.
Comparison of the low-order parameter values from Table 1 with the corresponding parameters determined by Kawashima et al. (2010) reveals relatively significant shifts in the values on a background of a general qualitative agreement (see Table B.1). Part of these shifts come from the basic difference in the models used, although both refer to the Rho-axis method. We follow the definition of the Rho-axis method given in Hougen et al. (1994) and, in our case, the A and E species are treated together with a single set of rotational parameters. On the contrary, Kawashima et al. (2010) treated the A and E species with distinct sets of rotational parameters (see their Table 2). As a result, a direct comparison of the changes in the rotational parameters is not possible. The same is true for the centrifugal distortion constants. At the same time, the change in V3 value is in good agreement with the prediction made in Kawashima et al. (2010) where a relatively large V6 term in the potential function was postulated by analogy with acetamide (Ilyushin et al. 2004). The data set available in Kawashima et al. (2010) was limited to the ground torsional state transitions only and did not give the opportunity to determine the V6 value. Thus, they examined how the V3 potential barrier is changed when a V6 term is added, assuming that the coupling of the vibrational modes of the NH group with the CH3 internal rotation has an effect similar to the one observed for the NH2 group in acetamide (Hirota et al. 2010). According to our results, the value of an index R used in Kawashima et al. (2010) to express the effect of the V6 term quantitatively is estimated to be 26.5 which is in a relatively good agreement with the value of 23.0 obtained in Kawashima et al. (2010) on the basis of ground state data only. Thus, our results support the general analysis of the CH3 internal rotation potential barrier in N-methylformamide provided in Kawashima et al. (2010).
A portion of the rotational spectrum of N-methylformamide measured around 133 GHz in the laboratory is shown in Fig. 2 and compared to the predicted rotational spectrum as provided by our current theoretical model. As can be seen from Fig. 2, the overall correspondence between experimental and theoretical spectra is very good. A slight inconsistency with intensity between predicted and observed spectra that may be visible for some strong lines is due to variations of source power and detector sensitivity.
 |
Fig. 2 Predicted (upper panel) and measured (lower panel) rotational spectrum of N-methylformamide between 133 and 133.5 GHz. The observed line shapes correspond to the first derivative of the actual line profile because frequency modulation and first harmonic lock-in detection are employed. The intensity axis is in arbitrary units. |
Predicted transitions of trans N-methylformamide in the vt = 0, 1, and 2 states.
The predictions of rotational transitions of trans N-methylformamide in the vt = 0, 1, and 2 torsionally excited states resulting from the fit are presented in Table 3. They are calculated for the frequency range up to 650 GHz and for the transitions with J ≤ 65. The table provides quantum numbers, followed by calculated transition frequencies and their uncertainties, the energy of the lower state and the product μ2S, where μ is the dipole moment of the molecule and S is the line strength of the transition. Owing to its significant size, the complete version of Table 3 is available at the CDS.
Partition functions of N-methylformamide and acetamide.
We provide the torsional-rotational (Qtr) and vibrational (Qv) partition functions of N-methylformamide in Table 4. The values of Qtr were calculated from first principles, that is, via direct summation over the rotational-torsional states. The maximum value of the J quantum number of the energy levels taken into account to calculate the partition function is 130, and excited torsional states up to νt = 8 were considered. The vibrational part, Qv, was estimated using a harmonic approximation and a simple formula that may be found in Gordy & Cook (1984, see their Eq. 3.60). The frequencies of the normal modes were obtained from DFT calculations of the harmonic force field using the B3LYP method and a 6-311++(3df, 2pd) basis set. Table 4 also lists the partition function values of acetamide that we calculated in a similar way as for N-methylformamide. The torsional-rotational part was calculated from first principales on the basis of the results presented in the paper by Ilyushin et al. (2004). To compute the Qv values, we used, when available, vibrational frequencies reported in the literature (Kutzelnigg & Mecke 1962; Kydd & Dunham 1980), but also the results of DFT calculations with the same method and basis set as for N-methylformamide. For both molecules, the values of Qv were calculated by taking all the vibrational modes into account except for the torsional mode which is already considered in Qtr. The full partition function, Qtot, is thus the product of Qtr and Qv.
4. Astronomical results
4.1. Observations
We use the full data set of the EMoCA spectral line survey obtained toward Sgr B2(N) with ALMA in its Cycles 0 and 1. The survey covers the frequency range between 84.1 and 114.4 GHz with a spectral resolution of 488.3 kHz (1.7 to 1.3 km s-1). The median angular resolution is 1.6″. A detailed description of the observations, the data reduction process, and the method used to identify the detected lines and derive column densities was presented in Belloche et al. (2016). Population diagrams are constructed in the same way as in our previous work. Here, we would like to emphasize the fact that the apparent discrepancy between the synthetic populations and the fit to the observed populations, the former lying below the latter (see, e.g., Fig 4b), results from the fact that the model is optimized to match (i.e., not overestimate) the peak temperatures of the detected transitions while the population diagram is based on integrated intensities. Because of the high level of line confusion, the wings of the detected lines are often still partially contaminated even after removing the contribution of the other known molecules included in our complete model. Therefore, the populations derived from the integrated intensities are, most of the time, slightly overestimated.
The complete model mentioned above refers to the synthetic spectrum that includes the emission of all the molecules that we have identified in Sgr B2(N2) so far (Belloche et al. 2013, 2014, 2016; Müller et al. 2016a,b; Margulès et al. 2016); it is overlaid in green in all figures that display observed spectra.
We focus our analysis on the secondary hot core Sgr B2(N2), which is located ~5″ to the North of the main hot core Sgr B2(N1) (Belloche et al. 2016). Sgr B2(N2) has narrower linewidths (FWHM ~ 5 km s-1) and thus exhibits a lower degree of line confusion than Sgr B2(N1). While some of the molecules reported below are also present on larger scales in the envelope of Sgr B2 (see, e.g., Jones et al. 2008), our interferometric observations are only sensitive to the compact emission arising from the embedded hot cores. As shown in Fig. 3, the emission analyzed in this section is compact and the derived column densities refer to the hot core Sgr B2(N2) only.
4.2. Tentative detection of N-methylformamide (CH3NHCHO)
We searched for emission lines of CH3NHCHO toward Sgr B2(N2) using the spectroscopic predictions obtained in Sect. 3. We compared the observed ALMA spectrum of Sgr B2(N2) to synthetic spectra of CH3NHCHO produced under the assumption of local thermodynamic equilibrium (LTE), which is expected to be valid given the high densities of the hot core regions probed by the EMoCA survey (Belloche et al. 2016). Figures A.1–A.3 show all the transitions of CH3NHCHO in its ground state and its first and second torsionally excited states that are covered by our survey and are expected to contribute significantly to the detected signal for typical hot core temperatures (150–200 K). In these figures, the synthetic spectrum containing the contribution of all molecules that we have identified toward Sgr B2(N2) so far, including CH3NHCHO, is overlaid in green on the observed spectrum, while the red spectrum shows the contribution of CH3NHCHO only, as derived from our best-fit LTE model. Most transitions of CH3NHCHO are blended with lines emitted by other molecules and therefore cannot be unambiguously assigned to CH3NHCHO. However, several lines are relatively free of contamination and match lines detected toward Sgr B2(N2), both in terms of linewidths and peak temperatures. These lines are marked with a star in Col. 11 of Tables B.2–B.4. In total, five lines can be clearly assigned to CH3NHCHO, four within its torsional ground state and one within its first torsionally excited state. Given the small number of clearly detected lines, we consider our detection of CH3NHCHO as tentative rather than secure.
Gaussian fits to the integrated intensity maps of four of the five detected lines indicate that the size of the emission is smaller than the beam. We obtain a size of ~1.8″ for the line at 93.41 GHz observed in setup S3 but this is also the setup with the worst angular resolution (2.9″ × 1.5″); we therefore do not fully trust this size. Indeed the other lines, at 91.89, 99.69 (see Fig. 3a), and 113.61 GHz, are all unresolved, suggesting a size smaller than 1″. We adopt a size of 0.9″ for our LTE modeling of CH3NHCHO. We did not attempt to fit a size to the integrated intensity map of the fifth detected line, at 111.22 GHz, because it is somewhat more contaminated on its low-frequency side.
 |
Fig. 3 Integrated intensity maps toward Sgr B2(N2) of a) CH3NHCHO 92,8–82,7, b) CH3NCO 101,10–91,9, c) NH2CHO ν12 = 140,4–30,3, d) NH213CHO 40,4–30,3, e) HNCO 43,1–33,0 and 43,2–33,1, and f) CH3CONH290,9–81,8, 91,9–81,8, 90,9–80,8, and 91,9–80,8. The position of Sgr B2(N2) is marked with a large cross. The smaller cross indicates the position of Sgr B2(N1). Because of the different systemic velocities of the two sources, the line assignment is only valid for Sgr B2(N2), inside the dashed box. The contours start at 4σ and increase by a factor of 2 at each step. The dashed (blue) contour is at −4σ. The value of the rms noise level, σ, is given in mJy beam-1 km s-1 in the top right corner of each panel. The mean frequency of the integration range in MHz and the synthesized beam are shown in the bottom right and left corners of each panel, respectively. |
 |
Fig. 4 Population diagram of CH3NHCHO toward Sgr B2(N2). The observed datapoints are shown in various colors (but not red) as indicated in the upper right corner of panel a) while the synthetic populations are shown in red. No correction is applied in panel a). In panel b), the optical depth correction has been applied to both the observed and synthetic populations and the contamination by all other species included in the full model has been removed from the observed datapoints. The purple line is a linear fit to the observed populations (in linear-logarithmic space). |
Rotational temperatures derived from population diagrams of selected complex organic molecules toward Sgr B2(N2).
The population diagram of CH3NHCHO is shown in Fig. 4. We used all detected lines plus a number of other ones that contribute significantly to detected lines and are contaminated by species that we have already identified and included in our full model. All the transitions used for the population diagram are listed in Tables B.2–B.4. Panel b of Fig. 4 shows the population diagram after correcting for the optical depth of the transitions, based on our best-fit LTE model of CH3NHCHO, and after removing the contamination by other species, based on our full model. A fit to this diagram yields a rotational temperature of 149 ± 20 K, with a significant uncertainty (Table 5). For the LTE modeling of the spectrum, we adopt a temperature of 180 K. The best-fit parameters are listed in Table 6.
In the following sections, we derive the column density of molecules that may be related to N-methylformamide in order to put the tentative detection of this molecule into a broader astrochemical context.
4.3. Methyl isocyanate (CH3NCO)
The first interstellar detection of methyl isocyanate was obtained toward Sgr B2(N) based on single-dish observations (Halfen et al. 2015, ; see also Cernicharo et al.2016), shortly after its in-situ detection in the frozen surface of comet 67P/Churyumov-Gerasimenko (Goesmann et al. 2015). Here, we use the predictions available in the Cologne database for molecular spectroscopy1 (CDMS, Müller et al. 2005) (tags 57 505 and 57 506, both version 1), which are based on measurements reported by Cernicharo et al. (2016) and Koput (1986).
Parameters of our best-fit LTE model of selected complex organic molecules toward Sgr B2(N2).
Methyl isocyanate is well detected toward Sgr B2(N2) in the EMoCA spectral survey: 60 lines are clearly detected in its vibrational ground state and four in its first vibrationally excited state (Figs. A.4 and A.5). Gaussian fits to the integrated intensity maps of the detected lines indicate a median emission size of ~1.2″ with a rms dispersion of ~0.1″ (see Fig. 3b). A fit to its population diagram shown in Fig. A.20 yields a rotational temperature of 141 ± 7 K (Table 5). We adopt a temperature of 150 K for our LTE modeling. The parameters of our best-fit model are given in Table 6. The model overestimates the peak temperature of a few transitions by ~30–40% (at 87.236, 96.062, 96.120, 104.793, and 104.856 GHz) while all other transitions are well fitted; the reason for these discrepancies is unclear.
4.4. Formamide (NH2CHO)
We use the CDMS entries for formamide in its ground and first vibrationally excited states (tags 45 512 and 45 516, versions 2 and 1, respectively) and for its 13C and 15N isotopologs in their ground state (tags 46 512 and 46 513, versions 2 and 1, respectively). These entries are based largely on Motiyenko et al. (2012), but also contain additional data. Laboratory data in the range of our survey were published by Kryvda et al. (2009). The entry for ν12 = 1 of the 13C isotopolog was prepared by one of us (HSPM) based on data from Stubgaard (1978).
Formamide is well detected toward Sgr B2(N2), with 30 lines in its vibrational ground state and 13 in its vibrationally excited state ν12 = 1 (Figs. A.6 and A.7). Its 13C isotopolog is also clearly detected, with 11 and 2 lines in its ν= 0 and ν12 = 1 states, respectively (Figs. A.8 and A.9). Finally, we report a tentative detection of 15NH2CHO, with one clearly detected line consistent with a 14N/15N isotopic ratio of 300 (Fig. A.10).
We derive a median emission size of ~0.9–1.0″ from Gaussian fits to the integrated intensity maps of the main and 13C isotopologs, with a rms dispersion of ~0.1″ (see Figs. 3c and d). A number of lines of the main isotopolog are saturated and not well fitted by our simple LTE model. We selected only the transitions with an optical depth lower than 2 to build the population diagram shown in Fig. A.21. A fit to this diagram yields a rotational temperature of 155 ± 3 K (Table 5). However, we obtain a rotational temperature of 200 ± 14 K when we limit the fit to the transitions that belong to the vibrational ground state. The population diagram of the 13C isotopolog is less populated (Fig. A.22) and yields a more uncertain rotational temperature of 222 ± 35 K (Table 5). We adopt a temperature of 200 K for our LTE model of all isotopologs of formamide.
We initially modeled the spectra assuming a size of 0.9″ as derived above but, in order to fit the ν12 = 1 transitions, we then had to assume a total column density of NH2CHO much lower than the one needed to fit the ground state transitions. By reducing the size to 0.8″, we could attenuate the discrepancy between ν= 0 and ν12 = 1, however we still need a total column density 1.4 times lower to fit ν12 = 1. This is surprising because we do not face this problem for the 13C isotopolog for which both states are well fitted assuming the same total column density. The discrepancy that affects the main isotopolog may be due to its higher optical depth although we do not feel that it is a satisfactory explanation.
4.5. Isocyanic acid (HNCO)
We use the CDMS entry for HNCO in its ground state (tag 43 511 version 1) by Lapinov et al. (2007) with additional measurements in the range of our survey by Hocking et al. (1975), and the JPL entry for the 13C isotopolog (tag 44 008 version 1, Hocking et al. 1975). Private entries for the vibrationally excited states ν5 = 1, ν6 = 1, and ν4 = 1 of the main isotopolog were prepared by one of us (HSPM). They are based on a preliminary, unpublished analysis of the ground and the four lowest excited states and were already used in Belloche et al. (2013). The data on the excited states was summarized in Niedenhoff et al. (1996). Transition frequencies in the range of our survey were published by Yamada & Winnewisser (1977) and Yamada (1977).
Isocyanic acid is also well detected toward Sgr B2(N2), with twelves lines in its vibrational ground state, four in its vibrationally excited state ν5 = 1, and one in its state ν6 = 1 (Figs. A.11–A.13). Its state ν4 = 1, is not unambiguously detected but contributes significantly to the signal detected at 87.97 GHz so we have included it in our full model (Fig. A.14). The 13C isotopolog is not unambiguously detected because all its significant transitions are located in the blueshifted wing of transitions of the main isotopolog, some of these suffering from absorption of the main isotopolog from the outer envelope of Sgr B2, which we have not yet taken into account in our full model (Fig. A.15). Nevertheless, the 13C isotopolog contributes significantly to the signal detected at several frequencies, so we have included it in our full model, based on the model derived below for the main isotopolog and assuming a 12C/13C isotopic ratio of 20.
We derive a median size of 0.9″ from Gaussian fits to the integrated intensity maps of the main isotopolog, with a dispersion of 0.2″ (see Fig. 3e). The population diagram shown in Fig. A.23 uses all but three transitions that are clearly detected, plus a number of additional lines that are more contaminated but for which we have identified and modeled the contaminating species. The three transitions of the vibrational ground state that we ignore (at 109.50, 109.91, and 110.30 GHz) have an opacity higher than 4. A fit to the population diagram yields a temperature of 220 ± 10 K. However, with this temperature, the optically thick lines of the ground state would saturate with a brightness temperature that is too low. As a compromise, we use a temperature of 240 K.
The detected lines of all four states of the main isotopolog are relatively well reproduced with the same model parameters (Table 6). A few issues remain, however. First of all, the peak temperatures of the optically thick lines of ν= 0 are slightly underestimated, except for the peak temperature of the 50,5–40,4 transition at 109.91 GHz which is overestimated, probably because of spatial filtering of extended emission not taken into account in our model (see Fig. 6 of Jones et al. 2008). The second issue concerns two transitions with an upper energy level Eup ~ 650 K, 341,34–350,35 at 85.37 GHz and 331,33–340,34 at 109.96 GHz, which are both overestimated. The former is located in a frequency range affected by c-C3H2 absorption by spiral arm clouds along the line of sight to Sgr B2, so the discrepancy at this frequency may not be a problem. We note, in addition, that the model for a third transition with Eup ~ 650 K, 54,1–44,0 at 109.78 GHz, is consistent with the detected signal. Therefore, it is unclear why our model overestimates the peak temperature of the transition at 109.96 GHz.
4.6. Acetamide (CH3CONH2)
We use predictions that are based on the measurements and analysis presented in Ilyushin et al. (2004) but were recomputed by one of us (V.V.I.) with the RAM36 code. We use the partition function calculated in Sect. 3 (Table 4).
With a total of 23 lines clearly detected in its ground state and its first and second torsionally excited states toward the hot core Sgr B2(N2), acetamide can be considered as securely detected (Figs. A.16–A.19). Fits to the integrated intensity maps of three of the detected lines give a median size of ~1.0″, with a dispersion of 0.2″ (see Fig. 3f). The emission of four other detected lines is unresolved, pointing to a smaller size. The maps of the remaining detected lines were not fitted because they are, to some level, contaminated in their wings, which could bias the size measurements. A fit to the population diagram yields a rotational temperature of 226 ± 33 K, which is not well constrained (Fig. A.24 and Table 5). We adopt a temperature of 180 K and a size of 0.9″ for our LTE model to make the comparison to N-methylformamide more straightforward. The detected lines of the ground state (νt = 0) and both torsionally excited states (νt = 1 and 2), as well as lines that connect different torsional states (Δνt ≠ 0), are well reproduced with the same model parameters (Table 6). One discrepancy can be noticed in Fig. A.16; the νt = 02520,6–2518,7 transition of the E species at 99.950 GHz does not have a counterpart in the observed spectrum (at the 3σ level). However it was extrapolated from a lower-J fit (J ≤ 20) and has a frequency uncertainty of 200 kHz which means that its frequency could be off by several times this number. Indeed, the actual positions of the hyperfine components of this line, as measured in the laboratory spectrum, are 99 951.872 MHz and 99 952.331 MHz, both with an uncertainty of 10 kHz. At these frequencies, strong emission is detected toward Sgr B2(N2), which reconciles the spectrum expected for acetamide with the observed spectrum. Therefore, the discrepancy between the observed and synthetic spectra at 99.950 GHz does not pose a significant problem.
5. Chemical modeling
To investigate the production of the tentatively detected N-methylformamide in Sgr B2(N2), we use the chemical kinetics model MAGICKAL (Garrod 2013) with an expanded gas-grain chemical network. This network is an extension of that presented by Belloche et al. (2014), and latterly by Müller et al. (2016a), and includes formation and destruction mechanisms for both CH3NHCHO and the related molecule CH3NCO. The model allows for a treatment of the fully-coupled gas-phase, grain/ice-surface, and ice-mantle chemistry. The physical model follows that detailed in previous papers where a cold collapse phase to maximum density (nH = 2 × 108 cm-3) and minimum dust-grain temperature (8 K) is followed by a warm-up from 8 to 400 K; during this phase, the gas and dust temperatures are assumed to be well coupled. The initial chemical compositions used in the model follow those of Garrod (2013). The reader is referred to the above-mentioned publications for a more detailed discussion of the basic physical and chemical model. In the models presented here, we use the intermediate warm-up timescale, which generally produces the best match between models and observed abundances of other chemical species. The warm-up model therefore reaches a temperature of 200 K at 2 × 105 yr, reaching 400 K (and the end of the model run) at ~2.85 × 105 yr.
The new network concentrates on the grain-surface production of the newly-introduced molecules. However, gas-phase destruction mechanisms for both molecules (as well as related intermediates) are included in the new network, the majority of which are ion-molecule processes or the subsequent dissociative recombination with electrons of the resultant molecular ions. Ion-molecule reactions are included for the major ionic species C+, He+, H3+, H3O+, and HCO+. Estimates for the rates of photo-dissociation of new molecules, as caused by cosmic ray-induced and (where extinction allows) external UV photons, are also included (see Garrod et al. 2008; Garrod 2013). Grain/ice-surface binding (desorption) energies for the new molecules are estimates based on interpolation/extrapolation of values for molecules with similar functional groups, following past publications. Binding energy values for CH3NCO and CH3NHCHO are 3575 K and 6281 K, respectively, based on the formulations [Edes(CH3) + Edes(N) + Edes(C) + Edes(O)] and [Edes(NH2CHO) - Edes(H) + Edes(CH3)]. We are not aware of any experimental determinations of these two quantities for appropriate surfaces.
In the new network, grain-surface and ice-mantle formation of CH3NCO occurs through a single radical-addition reaction: 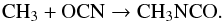 (7)Each of the necessary radicals may be formed either through repetitive atomic addition or through the photo-dissociation of or chemical H-abstraction from either CH4 or HNCO.
(7)Each of the necessary radicals may be formed either through repetitive atomic addition or through the photo-dissociation of or chemical H-abstraction from either CH4 or HNCO.
To form CH3NHCHO, a reaction involving the addition of a hydrogen atom to CH3NCO followed by the addition of another, presents itself as a possible route, that is,  The first of these two reactions requires the breaking of a carbon-nitrogen double bond, for which no activation energy barrier could be determined from the literature. The barrier to the similar reaction of H with HNCO has been determined experimentally in the gas phase by Nguyen et al. (1996) to be 1390 K, although the value for H + CH3NCO could plausibly be higher or lower. The expectation, however, is that, as with most barrier-mediated atomic-H reactions on cold grains, the mechanism would involve the tunneling of the H atom through the barrier, introducing further uncertainty into the reaction rate. By default, MAGICKAL uses a simple rectangular-barrier treatment to determine rates for tunneling reactions, typically assuming a uniform barrier width of 1 Å; the assumed height of the energy barrier therefore absorbs all other parameters pertaining to the overall reaction rate. The ideal activation energy barrier determined for the chemical model may therefore not be fully representative of the usual high-temperature value.
The first of these two reactions requires the breaking of a carbon-nitrogen double bond, for which no activation energy barrier could be determined from the literature. The barrier to the similar reaction of H with HNCO has been determined experimentally in the gas phase by Nguyen et al. (1996) to be 1390 K, although the value for H + CH3NCO could plausibly be higher or lower. The expectation, however, is that, as with most barrier-mediated atomic-H reactions on cold grains, the mechanism would involve the tunneling of the H atom through the barrier, introducing further uncertainty into the reaction rate. By default, MAGICKAL uses a simple rectangular-barrier treatment to determine rates for tunneling reactions, typically assuming a uniform barrier width of 1 Å; the assumed height of the energy barrier therefore absorbs all other parameters pertaining to the overall reaction rate. The ideal activation energy barrier determined for the chemical model may therefore not be fully representative of the usual high-temperature value.
Because a broad range of activation energy values could be plausible for reaction (8), in the chemical network, we initially assume that the rate for reaction (8) is negligible (models M1–M5), introducing non-zero values later.
Radical-addition reactions provide an alternative pathway to the formation of N-methylformamide, through the reactions: 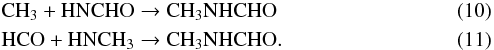 The larger radicals in each of the above two reactions are produced through the addition of NH to either CH3 or HCO. While cosmic ray-induced photo-dissociation of CH3NH2 and NH2CHO may also produce the necessary radicals, the chemical abstraction of a hydrogen atom by a radical from either molecule strongly favors the production of CH2NH2 or NH2CO, respectively, rather than the alternative radicals that could play a part in forming CH3NHCHO. However, in the case of H-atom abstraction from NH2CHO by OH, the model initially considers the barriers to NH2CO and NHCHO production to be similar (591 K versus 600 K), following the estimates used by Garrod (2013), although neither value has been determined rigorously. Consequently, the influence of the production of HNCHO through this mechanism is also examined in Sect. 5.1.
The larger radicals in each of the above two reactions are produced through the addition of NH to either CH3 or HCO. While cosmic ray-induced photo-dissociation of CH3NH2 and NH2CHO may also produce the necessary radicals, the chemical abstraction of a hydrogen atom by a radical from either molecule strongly favors the production of CH2NH2 or NH2CO, respectively, rather than the alternative radicals that could play a part in forming CH3NHCHO. However, in the case of H-atom abstraction from NH2CHO by OH, the model initially considers the barriers to NH2CO and NHCHO production to be similar (591 K versus 600 K), following the estimates used by Garrod (2013), although neither value has been determined rigorously. Consequently, the influence of the production of HNCHO through this mechanism is also examined in Sect. 5.1.
5.1. Model results
Model M1 comprises the initial model in which conversion of CH3NCO to CH3NHCHO via consecutive H addition (reactions (8) and (9)) is switched off.
 |
Fig. 5 Calculated abundances of selected chemical model species with respect to H2 for model M1, during warm-up from 8 K to 400 K. Solid lines indicate gas-phase abundances; dotted lines of the same color indicate solid-phase abundances of the same species. |
Figure 5 shows results from model M1. Time-dependent abundances for CH3NCO and CH3NHCHO as well as the related species HNCO, NH2CHO, and CH3CONH2 are shown. CH3NHCHO is seen to be formed on the grains (dotted line) in abundance at approximately 25 K, coincident with a significant growth in NH2CHO production. Its formation is dominated by reaction (10). CH3NCO production on the grains is much more modest, and occurs only through reaction (7). Its gas-phase abundance peaks at a relatively low temperature, following its desorption from grains, and falls again, although it later begins to rise as the abundant CH3NHCHO is photodissociated in the gas phase. CH3CONH2 reaches a peak abundance just a little larger than that of CH3NHCHO, forming mainly through the addition of CH3 and NH2CO radicals. HNCO is formed on the grains early (T< 20 K) via hydrogenation of OCN, which itself is formed through the atomic addition reactions O + CN and C + NO. The gas-phase abundance of HNCO peaks at close to 60 K as it desorbs from the grains, then falls away. It is a significant by-product of the destruction of larger molecules at later times in the model, and its abundance continues to grow until the final model temperature of 400 K is reached.
Peak gas-phase fractional abundances with respect to H2 and temperatures at which the peak abundance values are achieved, for models with varying activation energy barriers for the grain-surface reaction NH + CO → HNCO.
Table 7 shows the peak gas-phase abundances of the plotted species, along with the temperatures at which those peaks are reached. It may be noted that the peak abundance of CH3NHCHO is approximately at parity with HNCO, contrary to the observed column densities shown in Table 6, where a ratio of ~1:20 is obtained. The amount of CH3NHCHO produced in this model as compared with CH3NCO also appears high, while the abundance of CH3NCO is approximately one order of magnitude too low compared with HNCO. Since the sole production mechanism of CH3NCO relies on OCN, we have also considered alternative models, in which HNCO, a possible precursor to OCN, may be more easily formed.
Models M2–M5, whose results are also tabulated in Table 7, allow the grain-surface reaction NH + CO → HNCO to occur with a lower activation energy barrier than the 2500 K first assumed by Garrod et al. (2008), an estimate based loosely on the typically assumed barrier to the H-addition reaction H + CO → HCO. These models show two types of behavior, with the threshold falling somewhere between models M2 and M3, that is, an activation energy, EA, between 2000 K and 1500 K. Below this threshold, the models move away from M1-type behavior and instead show significantly increased HNCO production on the grains, such that the desorption of HNCO into the gas phase produces the peak abundance for this molecule. Figure 6 plots abundances for model M4, for which an activation energy barrier to the NH + CO reaction of 1250 K is assumed. The peak abundance of CH3NCO is notably increased (see also Table 7) as a result of the greater HNCO abundance, which contributes to the production of OCN. Conversely, the abundance of CH3NHCHO is found to decrease markedly in models M3-M5 as the NH + CO reaction becomes competitive with the NH + HCO → NHCHO reaction at the key temperature (~25 K) at which CH3 mobility makes reaction (10) important to CH3NHCHO production. Models M3-M5 all reach within a factor of ~2 of the observed CH3NHCHO/CH3NCO abundance ratio.
Peak gas-phase fractional abundances with respect to H2 and temperatures at which the peak abundance values are achieved, for models based on model M4, that is, with an activation energy barrier of 1250 K for the grain-surface reaction NH + CO → HNCO.
To investigate the importance of H-addition to CH3NCO, we adjust the conditions assumed in model M4, taking a selection of activation energy barriers for reaction (8) to give a non-zero reaction rate. M4A assumes the same value as the reaction H + HNCO, while models M4B–E increase this value incrementally (see Table 8). We also switch off the H-abstraction reaction OH + NH2CHO → NHCHO, to test its influence on CH3NHCHO production.
Model M4A demonstrates an extreme degree of conversion of CH3NCO to CH3NHCHO that is not borne out by the observations, while the somewhat higher barrier to hydrogenation of model M4B improves the match to the detected CH3NHCHO/CH3NCO ratio (see also Table 9), albeit with a value greater than unity. Model M4C, as with M4, shows a modest dominance of CH3NCO over CH3NHCHO, in line with observations, and is similar to M4 in the quality of its overall match with observations. At the higher activation energies used in models M4D and M4E, the importance of reaction (8) diminishes. Here, the influence of the removal of the OH + NH2CHO → NHCHO reaction becomes apparent via comparison with model M4 (Table 7); the abundance of CH3NHCHO falls by two-thirds. For each of the models M4A–E, the behavior of HNCO, NH2CHO, and CH3CONH2 are little affected in comparison to model M4.
Note that particularly in the cases of CH3NCO and CH3CONH2, the temperatures at which peak abundances are attained are somewhat lower than the rotational temperatures obtained from the spectroscopic model fits to the observational data. This may be due to the imprecision of either or both of the binding energy estimates and the spectroscopic fits. The observed HNCO temperature is best represented by the late-time/high-temperature peak found in the models, rather than the brief gas-phase spike at around 60 K. The precise temperature at which the high-temperature peak is reached is dependent on the destruction rates and warm-up timescale assumed in the model. In the case of CH3NCO, the larger spatial extent of this molecule (1.2”), as derived from the observations, compared with that of CH3NHCHO (0.9”) is consistent with the idea that the former is released from grains at lower temperatures than the latter.
Relative abundances for a selection of chemical species, from the initial model (M1) and the three other most successful models, and from the observations toward Sgr B2(N2)
Table 9 shows ratios of the abundances of the main molecules to HNCO, as well as the CH3NHCHO/CH3NCO and CH3NHCHO/CH3CONH2 ratios. Values are shown for the initial model M1, as well as representative models from Tables 7 and 8 that show a good match with observed values. Models M4 and M4C both show similarly good agreement with observed ratios involving CH3NCO and CH3NHCHO, while M4B is significantly further from the observed CH3NHCHO/HNCO ratio. However, all four models produce an overabundance of NH2CHO and CH3CONH2 by more than an order of magnitude. Because acetamide, CH3CONH2, is predominantly formed as a result of H-abstraction from formamide, followed by methyl-group addition, its overabundance is related to that of NH2CHO. The majority of the latter molecule is formed via the reaction NH2 + H2CO → NH2CHO + H, which we include on the grains as well as in the gas phase, assuming the activation energy barrier determined by Barone et al. (2015) of 26.9 K. However, Song & Kästner (2016) find a substantially higher barrier that would render the reaction rate negligible (as determined for a gas-phase interaction). In order to test this possibility, we remove the NH2+H2CO mechanism both in the gas phase and on grains. This reduces NH2CHO production by approximately an order of magnitude and brings the peak NH2CHO:HNCO ratio achieved in the models to a very good match with the observed value (1.2 versus 1.8). The removal of either the gas-phase or grain-surface mechanism alone is not sufficient to reduce NH2CHO abundance, as both contribute significantly in the present implementation. However, even the better match to observed NH2CHO is not sufficient to bring down acetamide abundances to appropriate levels, as the alternative NH2 + CH3CO formation mechanism is also important to its formation. The production of acetamide in hot cores merits further detailed study.
While models M4 and M4C show similar agreement with observations in spite of the different dominant chemical pathways involved for CH3NHCHO production, the fact that NH2CO is a more likely product of the reaction between OH and NH2CHO than NHCHO (judging by typical barriers to H-abstraction from an amino versus a carbonyl group) makes model M4C more plausible in this respect. The observed CH3NHCHO/CH3NCO ratio is bracketed by the values obtained with models M4B and M4C, indicating indeed that a mechanism of direct hydrogenation from CH3NCO to CH3NHCHO is capable of reproducing observations of these molecules. However, given an appropriate activation energy barrier, both reactions (8) and (10) may produce sufficient quantities of CH3NHCHO to agree reasonably with observations.
In order to achieve observed abundances of CH3NCO, a barrier to the reaction NH + CO → HNCO of no more than ~1500 K is required, allowing this to become the dominant mechanism by which HNCO is formed on grains (although its abundance in the gas phase is still dominated by its formation as a by-product of the destruction of larger species). Under such conditions, a barrier to the hydrogenation of CH3NCO may be estimated to be approximately 2000 K to best reproduce observed abundances of N-methylformamide.
6. Discussion
The spectroscopic results obtained in Sect. 3 represent a significant improvement in the characterization of the spectrum of N-methylformamide. First of all, we have substantially expanded the frequency coverage. Second, the rotational lines belonging to the first and second excited torsional states of N-methylformamide have been assigned and fitted for the first time. Finally, the resulting best model is capable of reproducing the assigned data set within experimental error, including the lines from previous studies that were excluded from the fits due to large observed-minus-calculated values (Kawashima et al. 2010).
All the chemical models computed in the course of this work predict a substantial abundance of CH3NHCHO compared to CH3NCO. The smallest ratios were produced by models M4D and M4E, with [CH3NHCHO]/[CH3NCO] ~ 0.07–0.09, but all other models have [CH3NHCHO]/[CH3NCO] ≳ 0.2. Two of the chemical models provide values that bracket the observed ratio, thus a fine tuning of the barrier against hydrogenation of CH3NCO would allow the observed ratio to be achieved. The chemical modeling thus gives some additional support to the tentative interstellar detection of CH3NHCHO.
N-methylformamide, CH3NHCHO, is a structural isomer of acetamide, CH3CONH2. As mentioned in Sect. 1, CH3NHCHO is the second most stable C2H5NO isomer, CH3CONH2 being the most stable one (Lattelais et al. 2010). In Sect. 4, we found that, provided its detection is confirmed, N-methylformamide is slightly less abundant than acetamide in Sgr B2(N2), which, at first sight, appears to be in agreement with the minimum energy principle initially put forward by Lattelais et al. (2009). This principle states that the most abundant isomer of a given generic chemical formula should be the most stable one thermodynamically. Lattelais et al. (2009) found a correlation between the observed abundance ratios of isomers of several generic chemical formulae as a function of their zero-point energy difference. According to this relation, N-methylformamide should be approximately 3.5 times less abundant than acetamide while it is only a factor 1.4 less abundant in Sgr B2(N2). The discrepancy is only slightly larger than a factor of two, but it tends to suggest that N-methylformamide does not follow this correlation closely. In addition, the range of kinetic parameters explored in the chemical models presented here produce variations in the abundance of CH3NHCHO of more than one order of magnitude. In the case of acetamide, while the models all produce an excess over the abundance of N-methylformamide, variations in other model parameters within a plausible range can produce variations in CH3CONH2 abundance that are comparable with those of CH3NHCHO. The availablility of key precursor radicals at the optimal temperature for diffusion is one of the key influences on the production of such molecules, and one that is unlikely to be controlled purely by the thermodynamic properties of the products. Therefore, the abundance ratio of the two isomers has most likely an origin based on kinetics, rather than a simple thermal equilibrium. A similar conclusion was obtained by Loomis et al. (2015) and Loison et al. (2016) based on observations of the isomers of C3H2O, whose abundance ratios are not consistent with the minimum energy principle.
A further test would be to measure the abundance of the next C2H5NO isomer, acetimidic acid, CH3C(OH)NH, which has a slightly higher zero-point energy than N-methylformamide (Lattelais et al. 2010). This would require laboratory measurements to characterize the rotational spectrum of this molecule and produce spectroscopic predictions suitable for an astronomical search. The dipole moment of this molecule, however, is more than a factor of 2 smaller than the ones of acetamide and N-methylformamide (Lattelais et al. 2009), making its detection more challenging.
7. Conclusions
The rotational spectrum of the trans conformer of N-methylformamide was studied in the laboratory in the frequency range from 45 to 630 GHz using two different spectrometers in Kharkiv and Lille. The new data provides significant expansion both in frequency range (from 118 GHz to 630 GHz) and quantum number coverage (from J = 11 to J = 62), including the first assignment of the rotational spectra of the first and second excited torsional states of N-methylformamide. The final data set contains 12 456 A- and E-type transitions in the ground, first, and second excited torsional states of the trans conformer. Our theoretical model fits the available data with a weighted root-mean-square deviation of 0.84, that is, within experimental error. The obtained results provide a firm basis for reliable predictions of the N-methylformamide spectrum in the millimeter and submillimeter wavelength range for the needs of radio astronomy.
Using the spectroscopic predictions obtained here, we report the first tentative interstellar detection of N-methylformamide. The main results of this study can be summarized as follows:
-
1.
Five transitions of N-methylformamide are coincident withspectral lines detected toward the hot molecular coreSgr B2(N2). These lines are not contaminated byother species, and their intensities are well reproduced by our LTEmodel of N-methylformamide. This suggests that the moleculemay be present in this source.
-
2.
We derive a column density of ~1 × 1017 cm-2 for N-methylformamide. The molecule is more than one order of magnitude less abundant than formamide, twice less abundant than methyl isocyanate, and only slightly less abundant than acetamide.
-
3.
Our gas-grain chemical kinetics model is able to reproduce the abundance ratio of N-methylformamide to methyl isocyanate using kinetic parameters within a plausible range, supporting the tentative detection of the former.
-
4.
The chemical models indicate that the efficient formation of HNCO via NH + CO on grains is a necessary step in the achievement of the observed gas-phase abundance of CH3NCO.
-
5.
Production of CH3NHCHO may plausibly occur on grains either through the direct addition of functional-group radicals or through the hydrogenation of CH3NCO.
-
6.
We also report the tentative detection of the 15N isotopolog of formamide toward Sgr B2(N2) with a 14N/15N isotopic ratio of 300.
Provided the detection of N-methylformamide is confirmed, the only slight underabundance of this molecule compared to its more stable structural isomer and the sensitivity of the model abundances to the variations of the model parameters suggest that the formation of these two molecules is controlled by kinetics rather than thermal equilibrium. The interstellar detection of the next stable isomer of the C2H5NO family, CH3C(OH)NH, may therefore become possible once its rotational spectrum has been measured in the laboratory.
Acknowledgments
This paper makes use of the following ALMA data: ADS/JAO.ALMA#2011.0.00017.S, ADS/JAO.ALMA#2012.1.00012.S. ALMA is a partnership of ESO (representing its member states), NSF (USA), and NINS (Japan), together with NRC (Canada), NSC and ASIAA (Taiwan), and KASI (Republic of Korea), in cooperation with the Republic of Chile. The Joint ALMA Observatory is operated by ESO, AUI/NRAO, and NAOJ. The interferometric data are available in the ALMA archive at https://almascience.eso.org/aq/. Part of this work was done within the Ukrainian-French CNRS-PICS # 6051 project. This work has been in part supported by the Deutsche Forschungsgemeinschaft (DFG) through the collaborative research grant SFB 956 “Conditions and Impact of Star Formation”, project area B3. Our research benefited from NASA’s Astrophysics Data System (ADS).
References
- Alekseev, E., Motiyenko, R., & Margulès, L. 2012, Radio Physics and Radio Astronomy, 3, 75 [NASA ADS] [CrossRef] [Google Scholar]
- Barone, V., Latouche, C., Skouteris, D., et al. 2015, MNRAS, 453, L31 [Google Scholar]
- Belloche, A., Müller, H. S. P., Menten, K. M., Schilke, P., & Comito, C. 2013, A&A, 559, A47 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Belloche, A., Garrod, R. T., Müller, H. S. P., & Menten, K. M. 2014, Science, 345, 1584 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Belloche, A., Müller, H. S. P., Garrod, R. T., & Menten, K. M. 2016, A&A, 587, A91 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Cernicharo, J., Kisiel, Z., Tercero, B., et al. 2016, A&A, 587, L4 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fantoni, A., & Caminati, W. 1996, J. Chem. Soc., Faraday Trans., 92, 343 [CrossRef] [Google Scholar]
- Fantoni, A. C., Caminati, W., Hartwig, H., & Stahl, W. 2002, J. Mol. Struct., 612, 305 [NASA ADS] [CrossRef] [Google Scholar]
- Garrod, R. T. 2013, ApJ, 765, 60 [Google Scholar]
- Garrod, R. T., Widicus Weaver, S. L., & Herbst, E. 2008, ApJ, 682, 283 [NASA ADS] [CrossRef] [Google Scholar]
- Goesmann, F., Rosenbauer, H., Bredehöft, J. H., et al. 2015, Science, 349, aab0689 [Google Scholar]
- Gordy, W., & Cook, R. L. 1984, Microwave Molecular Spectra, Techniques of Chemistry, Vol. XVIII (New York: Wiley) [Google Scholar]
- Halfen, D. T., Ilyushin, V., & Ziurys, L. M. 2011, ApJ, 743, 60 [NASA ADS] [CrossRef] [Google Scholar]
- Halfen, D. T., Ilyushin, V. V., & Ziurys, L. M. 2015, ApJ, 812, L5 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Hirota, E., Kawashima, Y., Usami, T., & Seto, K. 2010, J. Mol. Spectr., 260, 30 [NASA ADS] [CrossRef] [Google Scholar]
- Hocking, W. H., Gerry, M. C. L., & Winnewisser, G. 1975, Can. J. Phys., 53, 1869 [NASA ADS] [CrossRef] [Google Scholar]
- Hollis, J., Lovas, F. J., Remijan, A. J., et al. 2006, ApJ, 643, L25 [Google Scholar]
- Hougen, J. T., Kleiner, I., & Godefroid, M. 1994, J. Mol. Spectr., 163, 559 [NASA ADS] [CrossRef] [Google Scholar]
- Ilyushin, V. V., Alekseev, E. A., Dyubko, S. F., Kleiner, I., & Hougen, J. T. 2004, J. Mol. Spectr., 227, 115 [NASA ADS] [CrossRef] [Google Scholar]
- Ilyushin, V. V., Kisiel, Z., Pszczólkowski, L., Mäder, H., & Hougen, J. T. 2010, J. Mol. Spectr., 259, 26 [NASA ADS] [CrossRef] [Google Scholar]
- Ilyushin, V. V., Endres, C. P., Lewen, F., Schlemmer, S., & Drouin, B. J. 2013, J. Mol. Spectr., 290, 31 [NASA ADS] [CrossRef] [Google Scholar]
- Jones, P. A., Burton, M. G., Cunningham, M. R., et al. 2008, MNRAS, 386, 117 [NASA ADS] [CrossRef] [Google Scholar]
- Kaiser, R., Stockton, A., Kim, Y., Jensen, E., & Mathies, R. 2013, ApJ, 765, 111 [NASA ADS] [CrossRef] [Google Scholar]
- Kawashima, Y., Usami, T., Suenram, R. D., Golubiatnikov, G. Y., & Hirota, E. 2010, J. Mol. Spectr., 263, 11 [NASA ADS] [CrossRef] [Google Scholar]
- Kleiner, I. 2010, J. Mol. Spectr., 260, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Koput, J. 1986, J. Mol. Spectr., 115, 131 [NASA ADS] [CrossRef] [Google Scholar]
- Kryvda, A. V., Gerasimov, V. G., Dyubko, S. F., Alekseev, E. A., & Motiyenko, R. A. 2009, J. Mol. Spectr., 254,28 [NASA ADS] [CrossRef] [Google Scholar]
- Kutzelnigg, W., & Mecke, R. 1962, Spectrochim. Acta, 18, 549 [NASA ADS] [CrossRef] [Google Scholar]
- Kydd, R. A., & Dunham, A. R. C. 1980, J. Mol. Struct., 69, 79 [NASA ADS] [CrossRef] [Google Scholar]
- Lapinov, A. V., Golubiatnikov, G. Y., Markov, V. N., & Guarnieri, A. 2007, Astron. Lett., 33, 121 [NASA ADS] [CrossRef] [Google Scholar]
- Lattelais, M., Pauzat, F., Ellinger, Y., & Ceccarelli, C. 2009, ApJ, 696, L133 [Google Scholar]
- Lattelais, M., Pauzat, F., Ellinger, Y., & Ceccarelli, C. 2010, A&A, 519, A30 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Lin, C. C., & Swalen, J. D. 1959, Rev. Mod. Phys., 31, 841 [NASA ADS] [CrossRef] [Google Scholar]
- Loison, J.-C., Agúndez, M., Marcelino, N., et al. 2016, MNRAS, 456, 4101 [Google Scholar]
- Loomis, R. A., McGuire, B. A., Shingledecker, C., et al. 2015, ApJ, 799, 34 [Google Scholar]
- Margulès, L., Belloche, A., Müller, H. S. P., et al. 2016, A&A, 590, A93 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Motiyenko, R., Tercero, B., Cernicharo, J., & Margulès, L. 2012, A&A, 548, A71 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Müller, H. S. P., Schlöder, F., Stutzki, J., & Winnewisser, G. 2005, J. Mol. Struct., 742, 215 [NASA ADS] [CrossRef] [Google Scholar]
- Müller, H. S. P., Belloche, A., Xu, L.-H., et al. 2016a, A&A, 587, A92 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Müller, H. S. P., Walters, A., Wehres, N., et al. 2016b, A&A, 595, A87 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Nguyen, M. T., Sengupta, D., Vereecken, L., Peeters, J., & Vanquickenborne, L. G. 1996, J. Phys. Chem., 100, 1615 [CrossRef] [Google Scholar]
- Niedenhoff, M., Yamada, K. M. T., & Winnewisser, G. 1996, J. Mol. Spectr., 176, 342 [NASA ADS] [CrossRef] [Google Scholar]
- Rubin, R., Swenson Jr, G., Benson, R., Tigelaar, H., & Flygare, W. 1971, ApJ, 169, L39 [NASA ADS] [CrossRef] [Google Scholar]
- Song, L., & Kästner, J. 2016, Phys. Chem. Chem. Phys., 18, 29278 [NASA ADS] [CrossRef] [Google Scholar]
- Stubgaard, M. 1978, Ph.D. Thesis, Københavns Universitet, Denmark [Google Scholar]
- Xu, L.-H., Fisher, J., Lees, R., et al. 2008, J. Mol. Spectr., 251, 305 [NASA ADS] [CrossRef] [Google Scholar]
- Yamada, K. 1977, J. Mol. Spectr., 68, 423 [NASA ADS] [CrossRef] [Google Scholar]
- Yamada, K., & Winnewisser, M. 1977, J. Mol. Spectr., 68, 307 [NASA ADS] [CrossRef] [Google Scholar]
- Zakharenko, O., Motiyenko, R. A., Margulès, L., & Huet, T. R. 2015, J. Mol. Spectr., 317, 41 [Google Scholar]
Appendix A: Complementary figures
Figures A.1–A.19 show the transitions of CH3NHCHO, CH3NCO, NH2CHO, HNCO, CH3CONH2, and some of their isotopologs or vibrationally excited states that are covered by the EMoCA survey and contribute significantly to the signal detected toward Sgr B2(N2). Figures A.20–A.24 show the population diagrams of these molecules except the one of CH3NHCHO, which is shown in Fig. 4.
 |
Fig. A.1 Transitions of CH3NHCHO, νt = 0 covered by our ALMA survey. The best-fit LTE synthetic spectrum of CH3NHCHO, νt = 0 is displayed in red and overlaid on the observed spectrum of Sgr B2(N2) shown in black. The green synthetic spectrum contains the contributions of all molecules identified in our survey so far, including the species shown in red. The central frequency and width are indicated in MHz below each panel. The angular resolution (HPBW) is also indicated. The y-axis is labeled in brightness temperature units (K). The dotted line indicates the 3σ noise level. |
 |
Fig. A.4 continued. |
 |
Fig. A.4 continued. |
 |
Fig. A.6 continued. |
 |
Fig. A.16 continued. |
 |
Fig. A.16 continued. |
 |
Fig. A.16 continued. |
 |
Fig. A.17 continued. |
 |
Fig. A.17 continued. |
 |
Fig. A.18 continued. |
Appendix B: Complementary tables
Table B.1 provides the direct comparison between low-order spectral parameters determined in this study and the corresponding parameters from Kawashima et al. (2010).
Tables B.2–B.4 list the transitions that are used to build the population diagram of CH3NHCHO (Fig. 4). The transitions that we consider as clearly detected individually, that is, that are above the 3σ level and do not suffer too much from contamination by other species, are marked with a star. Because there are only five such transitions, the assignment of the corresponding five detected lines to CH3NHCHO is only tentative (see Sect. 4.2).
Comparison of the main spectroscopic parameters with previous results.
Selection of lines of CH3NHCHO νt = 0 covered by the EMoCA survey of Sgr B2(N2).
Selection of lines of CH3NHCHO νt = 1 covered by the EMoCA survey of Sgr B2(N2).
Selection of lines of CH3NHCHO νt = 2 covered by the EMoCA survey of Sgr B2(N2).
All Tables
Molecular parameters of the trans conformer of N-methylformamide obtained with the RAM36 program.
Rotational temperatures derived from population diagrams of selected complex organic molecules toward Sgr B2(N2).
Parameters of our best-fit LTE model of selected complex organic molecules toward Sgr B2(N2).
Peak gas-phase fractional abundances with respect to H2 and temperatures at which the peak abundance values are achieved, for models with varying activation energy barriers for the grain-surface reaction NH + CO → HNCO.
Peak gas-phase fractional abundances with respect to H2 and temperatures at which the peak abundance values are achieved, for models based on model M4, that is, with an activation energy barrier of 1250 K for the grain-surface reaction NH + CO → HNCO.
Relative abundances for a selection of chemical species, from the initial model (M1) and the three other most successful models, and from the observations toward Sgr B2(N2)
Selection of lines of CH3NHCHO νt = 0 covered by the EMoCA survey of Sgr B2(N2).
Selection of lines of CH3NHCHO νt = 1 covered by the EMoCA survey of Sgr B2(N2).
Selection of lines of CH3NHCHO νt = 2 covered by the EMoCA survey of Sgr B2(N2).
All Figures
|
Fig. 1 Structure of trans (a) and cis (b) conformations of N-methylformamide. |
| In the text | |
|
Fig. 2 Predicted (upper panel) and measured (lower panel) rotational spectrum of N-methylformamide between 133 and 133.5 GHz. The observed line shapes correspond to the first derivative of the actual line profile because frequency modulation and first harmonic lock-in detection are employed. The intensity axis is in arbitrary units. |
| In the text | |
|
Fig. 3 Integrated intensity maps toward Sgr B2(N2) of a) CH3NHCHO 92,8–82,7, b) CH3NCO 101,10–91,9, c) NH2CHO ν12 = 140,4–30,3, d) NH213CHO 40,4–30,3, e) HNCO 43,1–33,0 and 43,2–33,1, and f) CH3CONH290,9–81,8, 91,9–81,8, 90,9–80,8, and 91,9–80,8. The position of Sgr B2(N2) is marked with a large cross. The smaller cross indicates the position of Sgr B2(N1). Because of the different systemic velocities of the two sources, the line assignment is only valid for Sgr B2(N2), inside the dashed box. The contours start at 4σ and increase by a factor of 2 at each step. The dashed (blue) contour is at −4σ. The value of the rms noise level, σ, is given in mJy beam-1 km s-1 in the top right corner of each panel. The mean frequency of the integration range in MHz and the synthesized beam are shown in the bottom right and left corners of each panel, respectively. |
| In the text | |
|
Fig. 4 Population diagram of CH3NHCHO toward Sgr B2(N2). The observed datapoints are shown in various colors (but not red) as indicated in the upper right corner of panel a) while the synthetic populations are shown in red. No correction is applied in panel a). In panel b), the optical depth correction has been applied to both the observed and synthetic populations and the contamination by all other species included in the full model has been removed from the observed datapoints. The purple line is a linear fit to the observed populations (in linear-logarithmic space). |
| In the text | |
|
Fig. 5 Calculated abundances of selected chemical model species with respect to H2 for model M1, during warm-up from 8 K to 400 K. Solid lines indicate gas-phase abundances; dotted lines of the same color indicate solid-phase abundances of the same species. |
| In the text | |
 |
Fig. 6 As in Fig. 5 for model M4. |
| In the text | |
|
Fig. A.1 Transitions of CH3NHCHO, νt = 0 covered by our ALMA survey. The best-fit LTE synthetic spectrum of CH3NHCHO, νt = 0 is displayed in red and overlaid on the observed spectrum of Sgr B2(N2) shown in black. The green synthetic spectrum contains the contributions of all molecules identified in our survey so far, including the species shown in red. The central frequency and width are indicated in MHz below each panel. The angular resolution (HPBW) is also indicated. The y-axis is labeled in brightness temperature units (K). The dotted line indicates the 3σ noise level. |
| In the text | |
 |
Fig. A.2 Same as Fig. A.1 for CH3NHCHO, νt = 1. |
| In the text | |
 |
Fig. A.3 Same as Fig. A.1 for CH3NHCHO, νt = 2. |
| In the text | |
 |
Fig. A.4 Same as Fig. A.1 for CH3NCO, νb = 0. |
| In the text | |
|
Fig. A.4 continued. |
| In the text | |
|
Fig. A.4 continued. |
| In the text | |
 |
Fig. A.5 Same as Fig. A.1 for CH3NCO, νb = 1. |
| In the text | |
 |
Fig. A.6 Same as Fig. A.1 for NH2CHO, ν= 0. |
| In the text | |
|
Fig. A.6 continued. |
| In the text | |
 |
Fig. A.7 Same as Fig. A.1 for NH2CHO, ν12 = 1. |
| In the text | |
 |
Fig. A.8 Same as Fig. A.1 for NH213CHO, ν= 0. |
| In the text | |
 |
Fig. A.9 Same as Fig. A.1 for NH213CHO, ν12 = 1. |
| In the text | |
 |
Fig. A.10 Same as Fig. A.1 for 15NH2CHO, ν= 0. |
| In the text | |
 |
Fig. A.11 Same as Fig. A.1 for HNCO, ν= 0. |
| In the text | |
 |
Fig. A.12 Same as Fig. A.1 for HNCO, ν5 = 1. |
| In the text | |
 |
Fig. A.13 Same as Fig. A.1 for HNCO, ν6 = 1. |
| In the text | |
 |
Fig. A.14 Same as Fig. A.1 for HNCO, ν4 = 1. |
| In the text | |
 |
Fig. A.15 Same as Fig. A.1 for HN13CO, ν= 0. |
| In the text | |
 |
Fig. A.16 Same as Fig. A.1 for CH3CONH2, νt = 0. |
| In the text | |
|
Fig. A.16 continued. |
| In the text | |
|
Fig. A.16 continued. |
| In the text | |
|
Fig. A.16 continued. |
| In the text | |
 |
Fig. A.17 Same as Fig. A.1 for CH3CONH2, νt = 1. |
| In the text | |
|
Fig. A.17 continued. |
| In the text | |
|
Fig. A.17 continued. |
| In the text | |
 |
Fig. A.18 Same as Fig. A.1 for CH3CONH2, νt = 2. |
| In the text | |
|
Fig. A.18 continued. |
| In the text | |
 |
Fig. A.19 Same as Fig. A.1 for CH3CONH2, Δνt ≠ 0. |
| In the text | |
 |
Fig. A.20 Same as Fig. 4 for CH3NCO. |
| In the text | |
 |
Fig. A.21 Same as Fig. 4 for NH2CHO. |
| In the text | |
 |
Fig. A.22 Same as Fig. 4 for NH213CHO. |
| In the text | |
 |
Fig. A.23 Same as Fig. 4 for HNCO. |
| In the text | |
 |
Fig. A.24 Same as Fig. 4 for CH3CONH2. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.