| Issue |
A&A
Volume 562, February 2014
|
|
|---|---|---|
| Article Number | A22 | |
| Number of page(s) | 9 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201322495 | |
| Published online | 31 January 2014 | |
Photochemistry of PAHs in cosmic water ice
The effect of concentration on UV-VIS spectroscopy and ionization efficiency
1 Sackler Laboratory for Astrophysics, Leiden Observatory, Leiden Universtiy, PO Box 9513, 2300 RA Leiden, The Netherlands
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2 NASA Ames Research Center, Space Science & Astrobiology Division, MS 245-6, Moffett Field, CA 94035, USA
Received: 16 August 2013
Accepted: 14 November 2013
Abstract
Context. Observations and models show that polycyclic aromatic hydrocarbons (PAHs) are ubiquitous in the interstellar medium. Like other molecules in dense clouds, PAHs accrete onto interstellar dust grains, where they are embedded in an ice matrix dominated by water. In the laboratory, mixed molecular ices (not containing PAHs) have been extensively studied using Fourier transform infrared absorption spectroscopy. Experiments including PAHs in ices have started, however, the concentrations used are typically much higher than the concentrations expected for interstellar ices. Optical spectroscopy offers a sensitive alternative.
Aims. We report an experimental study of the effect PAH concentration has on the electronic spectra and the vacuum UV (VUV) driven processes of PAHs in water-rich ices. The goal is to apply the outcome to cosmic ices.
Methods. Optical spectroscopic studies allow us to obtain in-situ and quasi real-time electronic solid state spectra of two prototypical PAHs (pyrene and coronene) embedded in water ice under VUV photoprocessing. The study is carried out on PAH:H2O concentrations in the range of 1:30 000 to pure PAH, covering the temperature range from 12 to 125 K.
Results. PAH concentration strongly influences the efficiency of PAH cation formation. At low concentrations, ionization efficiencies are over 60% dropping to about 15% at 1:1000. Increasing the PAH concentration reveals spectral broadening in neutral and cation PAH spectra attributed to PAH clustering inside the ice. At the PAH concentrations expected for interstellar ices, some 10 to 20% may be present as cations. The presence of PAHs in neutral and ion form will add distinctive absorption bands to cosmic ice optical spectra and this may serve as a tool to determine PAH concentrations.
Key words: astrochemistry / molecular processes / methods: laboratory: solid state / ISM: molecules
© ESO, 2014
1. Introduction
Polycyclic aromatic hydrocarbon (PAH) molecules are observed along many lines of sight through their UV pumped IR emission features in the 3.3−18 μm region (e.g. Draine & Li 2007; Tielens 2008, and references therein). However, within dense molecular clouds, volatile molecules freeze onto the low temperature (10 to 50 K) dust particles (e.g. Boogert et al. 2008, 2011; Ehrenfreund 2002; Van Dishoeck 2008, and references therein) forming ice mantles around the dust grains. PAH molecules are expected to take part in this process causing them to become embedded in the ice mantles (e.g. Bernstein et al. 1999; Bouwman et al. 2009, 2011a; Gudipati et al. 2003, and references therein). Over the past two decades, a handful of papers have attributed interstellar IR absorption features to PAHs frozen in the ices (e.g. Sellgren et al. 1995; Keane et al. 2001). The remarkable absence of PAH IR emission features from within molecular clouds is fully consistent with PAHs embedded in interstellar ices.
Careful observations coupled with dedicated laboratory experiments carried out for nearly 50 years have shown that the main constituent of interstellar ice is H2O, with lower but substantial fractions of other volatile molecules such as CO, CO2, CH3OH and NH3 (e.g. Boogert et al. 2008, 2011; Ehrenfreund 2002; Pontoppidan 2004; Sellgren et al. 1995; Van Dishoeck 2008). Observations together with laboratory and theoretical studies (e.g. Bauschlicher et al. 2008, 2009; Steglich et al. 2011, 2012; Tielens 2008, and references therein) indicate that the PAH molecules observed in the gas phase contain about 50−100 carbon atoms effectively storing about 10 to 20% of the cosmic carbon inventory. PAH abundances within the ice are estimated to be up to 2% with respect to H2O (Bouwman et al. 2011b, and references therein). Since PAHs are significantly larger than the known typical interstellar ice components and each PAH molecule contains at least an order of magnitude more carbon than other ice constituents, they are an extraordinarily important source of carbon in interstellar ices. Furthermore, as shown in many previous studies of non-PAH containing ices, in-situ energetic processing driven by e.g. UV photons, free atoms or electrons, results in a very extensive reaction network (Allodi et al. 2013; Fuchs et al. 2009; Öberg et al. 2009, and references therein). Also, PAH containing ices will be subject to photoprocessing, specifically Ly-α photons that are the result of H2 emission after excitation by cosmic rays (Van Dishoeck et al. 2006). It is therefore of fundamental importance to understand the role that PAHs play in water-rich ices both from a spectroscopic and chemical point of view.
To start providing this information, initial studies of solid state PAH-ice spectroscopy and Ly-α-dominated vacuum ultraviolet (VUV) photochemistry have focused on the behavior of single PAH types isolated in H2O, CO, NH3 or noble gas, low temperature matrices (Bernstein et al. 2005, 2007; Bouwman et al. 2009, 2010, 2011a; Cuylle et al. 2012; Gudipati et al. 2003, 2006; Guennoun et al. 2011; Sandford et al. 2004). These deliberate simplifications, focusing on single PAHs in a pure matrix environment, are not representative for realistic interstellar ices, but are an essential first step in characterizing spectroscopic features and understanding fundamental photochemical reaction networks unique to PAHs, particularly in relation to the dominant interstellar ice component, H2O. The initial work of PAH photochemistry in water ice and water-rich ice mixtures took advantage of the great sensitivity of mass spectrometry. These studies were not in-situ studies, but rather focused on the non-volatile residual products, showing that PAHs undergo side group additions with the photofragments of the ice constituents (Bernstein et al. 1999, 2002).
Later in-situ studies followed, using two different techniques, each with both strengths and limitations. The first technique uses FTIR (Fourier Transform Infra-Red) absorption spectroscopy to observe the behavior of PAHs via their typical vibrational modes. Studying the photochemistry of the PAH pyrene (or coronene) suspended in argon and H2O matrices (Guennoun et al. 2011), confirmed side group addition reactions upon VUV irradiation. However, when PAHs are frozen in water ice, the spectral overlap between PAH absorption features and ice matrix features, in combination with the relatively weak oscillator strengths of PAH vibrational transitions, requires high PAH concentrations in order to detect any PAH photoproduct bands. Even with this, at best, only a few of such bands can be detected and these are generally severely blended with very broad water features (Bernstein et al. 2007). The required concentrations are typically higher than ~1:100 PAH:H2O and therefore not representative for interstellar conditions. The second technique, UV-VIS (Ultraviolet-visible) absorption spectroscopy, was developed to overcome this limitation by taking advantage of strong PAH electronic transitions (Bouwman et al. 2009; Gudipati et al. 2003). Small molecules (like H2O, CO, NH3) have, in contrast to PAHs and their ions, weak or no transitions in this spectral range. Taken together, these properties make it possible to record very clean optical spectra where only the strong, unique, and well-defined transitions related to the PAHs and their photoproducts are present, enabling one to track the UV-driven processes in-situ and in quasi real-time. The higher transition strength also facilitates the use of lower, astrophysically more relevant concentrations of PAH molecules in the ice matrix (typically ~1:5000 PAH:H2O). UV-VIS (hereafter also called optical) spectroscopic studies of irradiated ices have shown a very rich PAH ionization chemistry that is strongly dependent on matrix composition and temperature (Bouwman et al. 2011a; Cuylle et al. 2012; Gudipati et al. 2003, 2006).
Reviewing the results of both methods, an interesting difference in observations appears. On the one hand, optical (low density/concentration) studies of PAHs in H2O ice irradiated with VUV photons show that significantly more than 50% of the neutral PAH molecules are readily converted into their cations while, on the other hand (higher density/concentration) IR studies show very limited PAH ionization. The greater sensitivity of optical spectroscopy to significantly lower PAH concentrations in interstellar ice analogs over IR spectroscopy make it ideally suited to investigate the effect PAH concentration plays in the spectroscopy and photochemistry of PAHs in water ice. Knowledge of the fundamental processes affected by PAH:H2O concentration is essential before one can understand the role PAH concentration plays in interstellar ice chemistry. Since this seems to involve the interplay between ion-driven versus radical-driven processes, PAH concentration may play a very critical role in the energetic processing of interstellar ices. A systematic experimental study of this effect is presented here followed by an assessment of the astronomical implications.
Different PAH:H2O ice concentrations and temperatures studied here and the corresponding PAH photoionization yields.
2. Experimental method
The measurements for this study are performed with OASIS, our Optical Absorption Setup for Ice Spectroscopy, described in detail by Allodi et al. (2013) and Bouwman et al. (2009). It consists of a high vacuum chamber (P < 3 × 10-7 mbar) inside which a MgF2 ice deposition window is suspended on the cold finger of a closed cycle helium cryostat. A Lakeshore 330 temperature controller holds the deposition window at the desired temperature in the range of 12 to 325 K using a resistive heater wound around the cold finger. The absolute temperature accuracy is better than 1 K.
The ice is grown on the cold window from H2O vapor provided from an external glass bulb. The H2O vapor is guided through a 6 mm diameter stainless steel tube ending at a distance of 20 mm perpendicular to the deposition window inside the chamber. Between the bulb and the deposition window, a leak valve is installed and used to control the H2O deposition rate. The thickness of the ice is measured by laser interference using a HeNe laser (632.8 nm). This system comprises a photodiode to measure the intensity of the laser beam reflected off the growing ice and deposition window. A difference in path length between both reflections causes optical interference as the ice thickness increases, effectively modulating the photodiode signal intensity by about 20% over time. This method of measuring ice thickness is routine (Bossa et al. 2012, 2013; Romanescu et al. 2010) and is, for our specific case, described by Bouwman et al. (2009). The ice thickness (t) is given by: 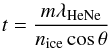 (1)With “m” being the amount of periods (or interference fringes) in the signal, λHeNe the wavelength of the HeNe laser, nice the refractive index of the ice (Romanescu et al. 2010) and θ the reflection angle, which in our case is 45 degrees. The density of amorphous solid water ice (Sceats & Rice 1983) combined with the ice thickness, allows one to determine the absolute column density of the matrix material. As the focus of the present study is on concentration effects, special care has been taken to perform these experiments as accurately as possible.
(1)With “m” being the amount of periods (or interference fringes) in the signal, λHeNe the wavelength of the HeNe laser, nice the refractive index of the ice (Romanescu et al. 2010) and θ the reflection angle, which in our case is 45 degrees. The density of amorphous solid water ice (Sceats & Rice 1983) combined with the ice thickness, allows one to determine the absolute column density of the matrix material. As the focus of the present study is on concentration effects, special care has been taken to perform these experiments as accurately as possible.
In this study, we embed coronene (Cor or C24H12) or pyrene (Py or C16H10), as prototypical PAHs in ice by co-deposition ensuring that the PAH:H2O ratio remains constant over the full depth of the ice. Coronene is sublimed in an oven consisting of a small metal cannister attached to the end of the deposition tube inside the vacuum chamber and is heated by a resistive wire. The temperature of the oven is controlled by setting the current through the resistive wire; the temperature is not directly measured. Since pyrene has a higher vapor pressure than coronene, a lower sublimation temperature suffices. It is sublimed using an external glass vial attached to the matrix deposition tube. The vial is heated to 30−80 °C using a wire heater. The temperature is measured using a type K thermocouple and controlled by modifying the current through the heater. This heater is also wound around the deposition tube to prevent pyrene condensation inside the deposition tube.
During deposition and photolysis, the PAH molecules and their photoproducts in the ice are monitored simultaneously in quasi real-time using an optical spectrometer (Andor Shamrock sr-303). The white light of a LOT-Oriel Xe arc lamp passes through two irises and is focused on the deposition window. After the deposition window, the light passes through a third iris and is refocused onto the entrance slit of the spectrometer using a lens. A 150 lines/mm grating disperses the light onto a CCD with 1024 pixels providing a spectral coverage from about 230 to 700 nm in a single go with a spectral resolution of 0.55 nm. During the measurements, multiple spectra are taken and averaged to improve signal to noise levels. In practice, a new spectrum is generated every 10 s. The first spectrum is taken as a reference (I0) to which all subsequent spectra are compared in units of optical depth (OD = ln(I / I0)). Besides measurement of the spectral data, the chamber pressure and window temperature are also recorded and stored.
The spectra measured before and after photolysis are used to determine the column density of the PAH and reaction products in the ice. For each spectrum, the band area of individual features is integrated and the resulting column density is derived (Kjaergaard et al. 2000), following; 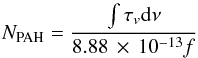 (2)where NPAH is the column density of the PAH molecules,
(2)where NPAH is the column density of the PAH molecules, 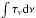 the integrated band area and f the oscillator strength of the involved transition. Knowing the column density of both the ice and the PAH molecules or eventual photoproducts allows their concentration to be obtained with a time resolution of 10 s. Taking the reproducibility of these experiments into account, we estimate an accuracy of about 10%. From these data, the maximum ionization yield is determined by averaging the cation values of 5 spectra around the maximum ionization yield.
the integrated band area and f the oscillator strength of the involved transition. Knowing the column density of both the ice and the PAH molecules or eventual photoproducts allows their concentration to be obtained with a time resolution of 10 s. Taking the reproducibility of these experiments into account, we estimate an accuracy of about 10%. From these data, the maximum ionization yield is determined by averaging the cation values of 5 spectra around the maximum ionization yield.
This study focuses on different concentrations of coronene (Aldrich, 97%):H2O (milli-Q, 3 freeze-pump-thaw cycles) deposited at 40 K, subsequently cooled or heated to 12, 25, 40, 80 or 125 K and VUV photoprocessed. A verification test with a similar concentration range is performed for pyrene (Aldrich, 99%):H2O (milli-Q, 3 freeze-pump-thaw cycles) deposited at 40 K and VUV processed at 12 K. The UV light is generated using a microwave powered H2 discharge lamp producing 1014 − 1015 photons cm-2 s-1 at the ice sample (Muñoz-Caro et al. 2002). The spectrum of the lamp consists mainly of Ly-α photons at 121.6 nm, but also a broadband emission centered around 160 nm.
Temperature dependent studies of PAH chemistry in water ice (Bouwman et al. 2011a; Gudipati et al. 2006) have shown that lower temperatures enhance the stability of PAH cations in the H2O ice matrix. Performing the measurements at 12 K as a reference provides maximal signal to noise levels of the observed cation features. Besides being the lowest temperature attainable using the OASIS setup, 12 K is also near the lowest temperatures observed for interstellar ices. Measurements with coronene at higher temperatures are also performed and serve as a consistency check.
2.1. Controlling PAH concentration
Ices are grown with the aim to realize PAH:H2O concentrations ranging from roughly 1:10 000, which is a typical value on the low end of the range for the OASIS setup up, to 1:100, the range where FTIR measurements are typically performed. The latter concentration is unusually high for the OASIS setup, requiring some challenges to be tackled. Increasing the column density of the PAH molecules in the ice has its limits as eventually a point is reached where the molecular absorption bands start to saturate, rendering the spectra useless. As a result of this limitation, the PAH concentration is mainly controlled through modifying the deposition rate of the H2O matrix, while keeping the deposition rate and column density of the PAH within set limits. Using this method, Py:H2O ices with concentrations in the range of 1:12 000 up to 1:80 have been successfully grown.
For ice thicknesses less than 400 nm, coronene presents a challenge as optical interference significantly deteriorates the baseline, degrading the reliability of the first series of spectra. Fortunately, this interference disappears as the ice becomes thicker. This effect does not occur when depositing very thin Py:H2O ices. A workaround for the high concentration coronene experiments, therefore, is realized by growing an ice in two steps. The first step is to grow a pure water ice with a thickness of about 500−600 nm. On top of this pure water ice, a very thin layer of ice with the desired high concentration of coronene is deposited. This eliminates the interference effect and makes it possible to grow ices at high Cor:H2O concentrations (1:1000 and higher). This method also allows one to accurately verify the deposition rate. It should be noted, though, that pores in 40 K deposited ice (Bossa et al. 2012, 2013) or other differences caused by the deposition method may also influence the overall outcome, but a direct comparison between both deposition methods with a ~1:1000 Cor:H2O ice does not reveal any substantial differences. Our measurements at 12, 25, 40, 80 and 125 K are performed at concentrations ranging from ~1:30 000 to ~1:120 Cor:H2O. Additionally, pure coronene is also deposited. An overview of all measurements is given in Table 1.
 |
Fig. 1 The 270−360 nm absorbance spectra of Cor:H2O ices at 5 different concentrations. All ices were deposited at 40 K. To aid comparison, all of the spectra are normalized to the peak of the S0–S3 transition. |
Oscillator strengths and transition assignments of the neutral and cation transitions of coronene and pyrene considered here.
3. Results
3.1. Coronene
The optical spectrum of coronene in H2O ice can be directly compared to earlier noble gas matrix work (Ehrenfreund et al. 1992) and the observations of Cor:H2O by Bouwman et al. (2011a). Figure 1 shows the 270 to 360 nm region of the spectrum of coronene in water ices at different concentrations ranging from 1:14 000 Cor:H2O to pure coronene, all deposited at 40 K. These spectra reveal two strong features at 300.5 nm and 337.0 nm, both accompanied by a multitude of weaker features, assigned by Ehrenfreund et al. (1992) to, respectively, the S0–S3 and S0–S2 transitions of neutral coronene. Increasing the concentration of coronene from 1:14 000 to 1:1100, 1:250, 1:120 Cor:H2O and eventually pure coronene results in progressive spectral broadening and a ~4 nm redshift for both transitions as well as a significant increase in relative intensity of the higher vibronic bands with respect to the 0−0 band for the S0–S3 transition.
In agreement with earlier observations (Bouwman et al. 2011a), photoprocessing of a ~1:14 000 Cor:H2O ice at 12 K instantly reveals – besides a sequence of decreasing neutral coronene bands – the growth of spectral features in the 350−475 nm range with the strongest new band at 463 nm (Fig. 2). All new features can be assigned to the coronene cation based on the work by Bouwman et al. (2011a); Ehrenfreund et al. (1992) and Shida (1988). As in previous work, the oscillator strength of the coronene cation is derived by assuming a 1:1 conversion from coronene neutral to coronene cation during the first 100 s of photolysis. However, although the same method was used, due to a difference in integration range, an ambiguity between the Ehrenfreund et al. (1992), Bouwman et al. (2011a) and this study exists where different oscillator strengths are obtained. The measurement made by Ehrenfreund et al. (1992) covered the spectrum from ~430 nm, missing the short wavelength wing observed by Bouwman et al. (2011a). Ehrenfreund et al. (1992) states a FWHM of 2.8 nm resulting in a very narrow integration range of roughly ~454.8 nm to ~463.2 nm. This resulted in an absolute oscillator strength of 0.012. Bouwman et al. (2011a) observed an extensive short wavelength wing related to the coronene cation and used the very broad integration range of 389 to 473 nm resulting in f = 0.23. We also observed the long wavelength wing, although in some of our measurements, the cation feature appeared very weak causing the short wavelength wing to be indistinguishable from the baseline. It was therefore chosen to limit the integration range of our measurements to only the strongest peak. In practice, this resulted in an integration range from 456 nm to 469 nm giving an oscillator strength of 0.0278, the value used throughout this study. These results are consistent, as Bouwman et al. (2011a) obtained the highest oscillator strength using the largest integration range (84 nm), followed by our study, utilizing a narrower integration range (13 nm) resulting in a smaller oscillator strength and finally, Ehrenfreund et al. (1992) who obtained the smallest oscillator strength using the smallest integration range (<~8, 4 nm). The relevant oscillator strengths are summarized in Table 2.
 |
Fig. 2 Absorbance spectrum (I) of coronene in H2O (1:14 000) at 12 K after ~12 min of vacuum UV photolysis referenced to the unphotolyzed spectrum (I0), shown at the bottom of Fig. 1. Negative peaks indicate the loss of neutral coronene while positive peaks indicate the appearance of new photoproducts, in this case the coronene cation. |
 |
Fig. 3 Growth of Cor+ and loss of Cor at different concentrations and different temperatures, as function of VUV dose (photolysis time). |
Normalization of all data with respect to the total deposited coronene column density for each experiment enables the direct comparison of the photo-dynamics between different experiments. Figure 3 shows the results for VUV irradiated Cor:H2O ice, illustrating the growth of coronene cation and loss of neutral coronene in the 12 (a−d), 40 (e, f) and 80 K (g, h) ices for different concentrations. The left column in Fig. 3 (measurements at 12 K) clearly shows the strong influence concentration has on the coronene cation production efficiency. Raising the PAH concentration substantially decreases the maximum cation yield. The plots in the right column of Fig. 3 shows that this trend holds in higher temperature ices as well; an interesting result since it has already been shown that for higher temperatures the ion production efficiency is lessened (Bouwman et al. 2011a). Comparing the measurements at 12 K (Fig. 3a to d) to those at 40 K (Fig. 3e and f) reveals no major differences between both measurement series, however, the measurements at 80 K (Fig. 3 panels g and h) show that the coronene cation is only barely present in the ice at a concentration 1:1200, where it is clearly more abundant for similar concentrations at 12 and at 40 K. This is consistent with the observations from Bouwman et al. (2011a) on the temperature dependence of the ionization yield of PAHs in water ice. The PAH ion stabilization lifetime is strongly dependent on temperature, with the highest yields found at the lowest temperatures (Bouwman et al. 2011a; Cuylle et al. 2012; Gudipati et al. 2006).
 |
Fig. 4 The 250−350 nm spectra of Py:H2O ices at 5 different concentrations. All ices were deposited at 40 K. |
 |
Fig. 5 Growth of Py+ and loss of Py at different concentrations and different temperatures, as function of VUV dose (photolysis time). |
3.2. Pyrene
The spectrum of neutral pyrene in a H2O matrix shows a strong feature at 334 nm which corresponds to the 0−0 band of the 1B2u, S0–S2 transition, accompanied by weaker vibronic bands on the short wavelength side (Fig. 4) (Bouwman et al. 2009). When comparing the pyrene spectra deposited at increasing concentrations in H2O, a slight broadening in combination with a small redshift becomes apparent (Fig. 4), however, both effects are less pronounced than for coronene. As with coronene, there is also an increase in relative intensity of higher vibronic bands with respect to the 0−0 band in only one of the two electronic transitions measured, but this time it occurs in the S0–S2 transition rather than the S0–S3 transition. Upon photolysis, a number of new features appear in the 350−470 nm range, most of which, can be assigned to the Py cation. This behavior has been extensively documented by Bouwman et al. (2010, 2011a) and Cuylle et al. (2012) and is not further elaborated here. The strongest pyrene cation band falls at 446 nm and has been assigned to the 2Au transition (Shida 1988). The oscillator strength of this Py cation band has been derived using the same method as described for coronene in Sect. 3.1. Assuming a 1:1 conversion from pyrene to the pyrene cation during the first 100 s of photolysis results in an oscillator strength of f = 0.104 for the 446 nm transition. In this case, band integration is performed from 409 to 460 nm. The maximum ionization yields for the concentrations studied here are listed in Table 1 and plotted in Fig. 6.
As with coronene, normalizing the spectra in each separate experiment to the total column density of the initially deposited neutral pyrene allows inter-comparison of the data from each experiment. Figure 5 shows the loss of neutral pyrene and growth of the pyrene cation upon VUV photolysis at 12 K for three different concentrations. The same overall trend as observed for coronene is apparent; the maximum cation ionization yield decreases as the pyrene concentration increases. However, there are also several small, but significant, differences between the ionization behavior of coronene and pyrene. In contrast with coronene, Py cation growth continues (albeit slowly) or stabilizes at long photolysis times whereas it drops earlier with coronene. Additionally, overall ionization seems to be more efficient at higher concentrations.
 |
Fig. 6 Maximum ionization yield for different temperatures and concentrations of pyrene and coronene in H2O matrix. |
3.3. Ionization efficiency
Figure 6 shows the maximum ionization yields for all coronene:H2O (and pyrene:H2O at 12 K) measurements plotted versus their respective concentrations as listed in Table 1. There are two distinct regimes visible. The first is a rapid drop from the lowest concentrations (PAH:H2O <1:10 000) to about 1:1000 PAH:H2O followed by a much slower (and fractionally smaller) drop with PAH:H2O above 1:1000. This illustrates, once again the concentration effect on solid PAH ionization efficiency.
4. Discussion
Understanding the photochemical processes at play in both the ice and liquid forms of water remains an active area of research in its own right (Chipman et al. 2011; Gillis et al. 2001; Marsalek et al. 2011; Verlet et al. 2005, and reference therein). In this section we restrict the discussion of the concentration effects presented above in terms of the factors that are likely to play dominant roles, namely PAH cluster formation, site geometry, ice micro-structure and PAH inter-molecular distance. Based on the overall, macroscopic physical and chemical PAH:H2O ice environment, a rationalization of the concentration dependent behavior shown in Fig. 6 is presented subdivided into a low concentration and a high concentration regime. It should be noted that we do not aim to describe our observations at a molecular physics level, as this lies beyond the scope of the present paper.
4.1. Low concentration regime (PAH:H2O < 1:1000)
The low concentration regime shows a drop in maximum ionization by half at concentrations of the order of 1:4000, a range where the optical spectra do not reveal features related to PAH clustering in the ice as can be seen in Figs. 1 and 4. The most dramatic impact of clustering on the electronic transitions occurs when the PAH:H2O ratio is well above 1:1000. This is fully consistent with an IR spectroscopic study of PAH clustering behavior for the PAH naphthalene isolated in an argon matrix (Roser & Allamandola 2010). They found the onset of weak spectroscopic effects due to clustering in the CH stretching and CH out-of-plane bending modes at ratios above 1:1000 and no evidence in the CC stretching region until concentrations exceed 1:500. Since these concentrations are far greater than those for which maximum PAH ionization drops precipitously, we conclude that PAH cluster formation is not responsible for the rapid decrease in maximum ionization yield with increasing PAH:H2O concentration as shown in Fig. 6. Furthermore, the maximum ionization yields for both coronene and pyrene show a similar overall concentration dependence. Spectroscopic evidence shows that site geometry only plays a role at the concentration range where PAH isolation is no longer possible (PAH:H2O is higher than 1:1000). The geometrical size of coronene and pyrene significantly differ, and therefore likely require different numbers of H2O molecules to isolate and stabilize their ion forms.
When asessing the influence of ice micro-structure and PAH concentration, it is important to recognize the critical role that water plays in interstellar ice astrochemistry e.g. (Öberg et al. 2010), overall ice structure and physical behavior (Bossa et al. 2012, 2013; Collings et al. 2005; Hawkins et al. 2005; Kumi et al. 2006; Malyk et al. 2007). The complex interplay between water molecules and charged small species embedded has been studied extensively using theoretical methods (Park & Woon 2004a,b; Woon 2004, and references therein). From these studies, it was found that the number and orientation of the water molecules influence the ionization energies, potentially enabling auto-ionization in interstellar ices not containing PAHs. Most of these studies have focused on species of sizes, polarizabilities and dipole moments similar to that of water (Woon 2006, and reference therein). PAHs are very different. They are much larger than water molecules, they have very large polarizabilities and pure PAHs have very small permanent dipole moments. The earlier experimental studies of PAHs in water ices demonstrated the ease and efficiency of PAH ionization and stabilization and the role of temperature on those processes. The resulting PAH ionization energy lowering, along with the concentration dependence described here show the critical and unique role water plays as none of these phenomena are observed in noble gas matrices.
In addition to the break up of H2O molecules into H and OH upon VUV irradiation, another important early step in PAH photolysis is: 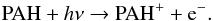 The electron affinity (EA) of water ice is estimated to be between 2 and 3 eV (Khan 2004; Novakovskaya & Stepanov 2004) and the EA of the OH radical in ice is computed to be 5.06 eV. As long as the PAH concentrations are low enough, VUV irradiated water-rich ices provide ample sites that can trap electrons in 5 eV wells (Woon & Park 2004). This, in combination with the added thermodynamic stability associated with the solvation energy of the isolated PAH cation in H2O ice enables the stabilization of PAH cations in these ices, rather than electron recombination and neutralization. This effect appears very pronounced when comparing PAH ionization yields between low concentration H2O matrices and low concentration noble gas matrices. The maximal ionization yield in H2O matrices is reported to be higher than 50%, while in noble gas matrices, they are typically only between 2 to 10% (Hudgins & Allamandola 1995, 1997).
The electron affinity (EA) of water ice is estimated to be between 2 and 3 eV (Khan 2004; Novakovskaya & Stepanov 2004) and the EA of the OH radical in ice is computed to be 5.06 eV. As long as the PAH concentrations are low enough, VUV irradiated water-rich ices provide ample sites that can trap electrons in 5 eV wells (Woon & Park 2004). This, in combination with the added thermodynamic stability associated with the solvation energy of the isolated PAH cation in H2O ice enables the stabilization of PAH cations in these ices, rather than electron recombination and neutralization. This effect appears very pronounced when comparing PAH ionization yields between low concentration H2O matrices and low concentration noble gas matrices. The maximal ionization yield in H2O matrices is reported to be higher than 50%, while in noble gas matrices, they are typically only between 2 to 10% (Hudgins & Allamandola 1995, 1997).
4.2. High concentration regime (PAH:H2O > 1:1000)
Increasing the concentration above 1:1000 PAH:H2O gradually reveals the spectral broadening and redshift in both the neutral and cation spectra (see Figs. 1 and 4). This phenomenon is related to PAH clustering in the ice. Indeed, increasing the PAH concentration reduces the average distance between PAH molecules, effectively increasing the recombination reaction channel and simultaneously decreasing the number of water related traps. In this reaction channel, an electron, freed upon VUV photolysis recombines with another PAH cation already present inside the ice matrix rather than undergoing a competing reaction channel. This is clearly more likely for concentrations in which the average distance between PAHs is small enough such that significant clustering occurs.
A further steady decrease of maximal ionization yield is observed until the point where a significant difference between both PAHs is apparent. Comparing the ionization fractions between pyrene and coronene, pyrene consistently has higher ionization fractions. The origin of this difference in behavior is not a priori clear as the effects of PAH clustering are not studied in detail. The type of clusters (parallel or perpendicular) and PAH size may play a role.
5. Astrophysical implications
PAH containing ices have not been identified in space yet. Infrared laboratory studies have been ambiguous, as different PAHs show similar spectra, because the typical vibrational modes involved are identical. However, unique electronic solid state spectra that have become available over the last years (Gudipati et al. 2003; Bouwman et al. 2011a; Cuylle et al. 2012) provide laboratory data to realize the observational identification of PAH species in the optical domain. The focus of the present study is the reactivity of PAH containing ice. As with the earlier studies of the fundamental processes driven by the VUV photolysis of PAHs in water ice, the concentration studies reported here have important implications to our understanding of ices in astronomical objects of different types, ranging from icy bodies in the solar system to the interstellar medium. This work presents the first detailed studies of the effect of concentration on PAH ionization efficiency in H2O ice. We show that there is a clear link between PAH concentration and photoionization yield and thus, by implication, with the types of initial PAH photoproducts present in the ice that are available for subsequent reactions and processes. Here we consider some of the possible implications regarding interstellar ices.
The behavior of ionization efficiency in H2O ice falls in two distinct concentration regimes. At low PAH concentrations, ionization yield and stabilization is high, ranging from ~70% at the lowest concentrations studied here to about 15 to 20% at PAH:H2O ~1:1000, and reducing to a few percent for Cor:H2O 1:250 and nearly 10% for Py:H2O 1:80. At higher concentrations, the drop off with increasing concentration gradually slows down, varying from 10 to 15% for both pyrene and coronene at 1:1000 reducing to no ionization for ~1:100 coronene. PAH concentrations with respect to H2O in interstellar ices are estimated on the order of up to 2−3% (e.g. Bouwman et al. 2011b; Keane et al. 2001; Sellgren et al. 1995). However, at this stage in our understanding, PAH concentration in interstellar ices can only be loosely defined and it is likely that the effective concentration from the perspective of the overall ice is lower. There are two main reasons behind this. First, interstellar PAH abundance is estimated from the intensity of the mid-IR emission features from mixtures of large PAHs (30 < C < 100) in the gas phase that are associated with UV rich regions. The PAHs studied here are much much smaller. Lower PAH concentrations in interstellar ices are required to satisfy the constraints imposed by cosmic C/H abundances. Second, there are other molecules in interstellar ices besides H2O. The relative abundances of the molecules known to be present in interstellar ices are not given in percentages because the full molecular make-up of interstellar ices is unknown. It is for this reason that lists of the species comprising interstellar (and likewise cometary ices) are given with respect to H2O, which is usually given a value of 100. Given typical interstellar ice compositions and relative abundances w.r.t. H2O (roughly H2O = 100; CO and CO2 ~ 30−50; CH3OH, H2CO ~ 20−30; NH3 ~ 5−15; etc.), it is clear that the total concentration of different PAHs in interstellar ices is very likely to be of the order of a few percent. We assume that the conclusions derived here for the concentrations of two individual PAHs can be used to describe the cumulative effect of a mixture of PAHs. Exact numbers, however, are not available. We do not want to exclude a priori a scenario in which the total PAH abundance is even lower, and in such an ultra-low domain we expect that a similar situation applies as for the processes described at low concentration, but less pronounced.
For the following, we conservatively adopt interstellar PAH:H2O ice concentrations in the 0.1−0.5% range i.e., 1:1000 to 1:200, percentages that fall in the high concentration regime. These concentrations are lower than the 2−3% PAH:H2O concentrations derived by Bouwman et al. (2011b) and consistent with the values implied from Sellgren et al. (1995) and Keane et al. (2001). In this concentration range, PAH ionization occurs, with conversion efficiencies on the order of 10−20%. Under these laboratory conditions, the maximum yield drops and further processing occurs. Under interstellar conditions, these processes are much slower. Combining the concentration dependence of the VUV processes described here with the astrophysical models of PAH freeze out and processing presented by Bouwman et al. (2011a), reinforces the conclusion that UV induced, radical driven processes such as PAH ionization processes could be important. The presence of ionized species within interstellar ices, or cosmic ices in general, and their roles in the chemical, physical and spectroscopic properties of these ices has not been seriously considered. Since ion-mediated processes are so fundamentally different from the reactions normally considered for cosmic ice chemistry such as neutral-neutral (radical) reaction networks, the role of PAH ions as well as other ionized species should be included when modeling these ices. Support for the importance of ions in cosmic ices is provided by the 2165 cm-1 interstellar ice absorption band associated with high-mass protostellar regions. After a long controversy in the literature, this band has been assigned to OCN− (Demyk et al. 1998; Novozamsky et al 2001; Schutte & Greenberg 1997, and references therein). However, a long-standing problem associated with this is the identity of the counter cation or cations. Given all the evidence, the presence of PAH cations in these ices, in combination with other cations, such as NH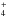 (Schutte & Khanna 2003), may provide part of the solution to this problem as well.
(Schutte & Khanna 2003), may provide part of the solution to this problem as well.
Another important result of this work is that a significant amount of spectral broadening and intensity changes are observed in the optical bands. Beyond the normal matrix induced band broadening and shifting, an electronic state dependent vibronic band intensity increase occurs with respect to the 0−0 transition, as PAH concentration increases. If this effect is a general trend with PAHs and PAH mixtures, this has important implications for the detection and identification of PAHs in interstellar ices with similar concentrations. In such ices, the neutral PAH spectra can be broadened and redshifted by a few nm, effectively smoothing the spectra. Once optical solid state PAH detections have become possible, which currently is not the case but definitely a goal to aim at, this may offer a diagnostic tool to derive also the concentration of a specific PAH. Observationally, this will be difficult as broad features are hard to separate from the underlying continuum. Also, given that many different frozen PAHs are present, spectral blending may further complicate such efforts. The laboratory data to realize this, however, are now (getting) available.
6. Conclusions
This work reports the concentration dependent behavior of the PAHs pyrene and coronene embedded in a H2O matrix under VUV photolysis at different temperatures (12, 25, 40, 80 and 125 K). The study is based on UV-VIS spectroscopic measurements of PAH:H2O samples with concentrations ranging from 1:30 000 to 1:80 irradiated with a Ly-α light source. Two PAHs are considered in order to obtain a wide overview of the processes influencing the photoionization efficiency. The oscillator strengths of the PAH+ electronic transitions have been derived and used in a time dependent study to track the abundance of the respective PAH cations in the ice. The main conclusions from the experiments are:
-
1.
PAH ionization in the water matrix upon VUV irradiation features two very distinct regimes governed by changing concentration. The first is a rapid drop from the lowest concentrations (PAH:H2O < 1:10 000) to about 1:1000 PAH:H2O, followed by a much slower (and fractionally smaller) drop with PAH:H2O above 1:1000. This behavior is observed at different temperatures and with different PAHs suggesting this may be a general effect amongst PAHs.
-
2.
At concentrations expected in interstellar ices, ~10−20% of the PAH may be present as cations perhaps serving as part of the cation population needed to balance the OCN− abundance.
-
3.
The spectra of PAHs embedded in H2O ice at concentrations of ~1:1000 and higher feature spectral broadening and redshift progressing with increased concentration. This effect is the result of the formation of small PAH clusters inside the ice matrix. In the interstellar medium where a multitude of PAH species are expected to be present, this effect might contribute to smoothing and blending of the optical absorption features, making PAH identification difficult.
Acknowledgments
This research is financially supported by the Netherlands School for Astronomy, NWO-VICI, the Dutch Organisation for Science, and the European Communitys 7th Framework Programme (FP7/2007-2013) under grant agreement n.238258. L.J.A. gratefully acknowledges support from NASA’s Laboratory Astrophysics and Astrobiology programs, NASA’s Laboratory Astrophysics “Carbon in the Galaxy” Consortium and NASA Ames Research Center’s Exchange program with the Netherlands.
References
- Allamandola, L. J., Hudgins, D. M., Bauschlicher, C. W., & Langhoff, S. R. 1999, A&A, 352, 659 [NASA ADS] [Google Scholar]
- Allodi, M. A., Baragiola, R. A., Baratta, G. A., et al. 2013, Space Sci. Rev., 180, 101 [NASA ADS] [CrossRef] [Google Scholar]
- Apostolopoulou, V., Vakros, J., Kordulis, C., & Lycourghiotis, A. 2009, Coll. Surf. A Phys. Chem. Eng. Aspects, 349, 189 [CrossRef] [Google Scholar]
- Bauschlicher, C. W., Peeters, E., & Allamandola, L. J. 2008, ApJ, 678, 316 [NASA ADS] [CrossRef] [Google Scholar]
- Bauschlicher, C. W., Peeters, E., & Allamandola, L. J. 2009, ApJ, 697, 311 [NASA ADS] [CrossRef] [Google Scholar]
- Bernstein, M. P., Sandford, S. A., Allamandola, L. J., et al. 1999, Science, 283, 1135 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Bernstein, M. P., Elsila, J. E., Dworkin, J. P., et al. 2002, ApJ, 576, 1115 [NASA ADS] [CrossRef] [Google Scholar]
- Bernstein, M. P., Sandford, S. A., & Allamandola, L. J. 2005, ApJS, 161, 53 [NASA ADS] [CrossRef] [Google Scholar]
- Bernstein, M. P., Sandford, S. A., Mattioda, A. L., & Allamandola, L. J. 2007, ApJ, 664, 1264 [NASA ADS] [CrossRef] [Google Scholar]
- Bito, Y., Shida, N., & Toru, T. 2000, Chem. Phys. Lett., 328, 310 [NASA ADS] [CrossRef] [Google Scholar]
- Boogert, A. C. A., Pontoppidan, K. M., Knez, C., et al. 2008, ApJ, 678, 985 [NASA ADS] [CrossRef] [Google Scholar]
- Boogert, A. C. A., Huard, T. L., Cook, A. M., et al. 2011, ApJ, 729, 92 [NASA ADS] [CrossRef] [Google Scholar]
- Bossa, J.-B., Isokoski, K., de Valois, M. S., & Linnartz, H. 2012, A&A, 545, A82 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bossa, J.-B., Isokoski, K., Paardekooper, D. M., et al. 2013, A&A, 561, A136 [Google Scholar]
- Bouwman, J., Paardekooper, D. M., Cuppen, H. M., Linnartz, H., & Allamandola, L. J. 2009, ApJ, 700, 56 [NASA ADS] [CrossRef] [Google Scholar]
- Bouwman, J., Cuppen, H. M., Bakker, A., Allamandola, L. J., & Linnartz, H. 2010, A&A, 511, A33 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bouwman, J., Cuppen, H. M., Steglich, M., Allamandola, L. J., & Linnartz, H. 2011a, A&A, 529, A46 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Bouwman, J., Mattioda, A. L., Linnartz, H., & Allamandola, L. J. 2011b, A&A, 525, A93 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Chipman, D. M. 2011, J. Phys. Chem. A, 115, 1161 [CrossRef] [Google Scholar]
- Collings, M. P., Dever, J. W., McCoustra, M. R. S., & Fraser, H. J. 2005, in Highlights of Astronomy, vol. 13 (ASP), 491 [Google Scholar]
- Cuylle, S. H., Tenenbaum, E. D., Bouwman, J., Linnartz, H., & Allamandola, L. J. 2012, MNRAS, 423, 1825 [NASA ADS] [CrossRef] [Google Scholar]
- Demyk, K., Dartois, E., D’Hendecourt, L., et al. 1998, A&A, 339, 553 [NASA ADS] [Google Scholar]
- Draine, B. T., & Li, A. 2007, ApJ, 657, 810 [NASA ADS] [CrossRef] [Google Scholar]
- Ehrenfreund, P. 2002, in Highlights of Astronomy, vol. 12 (ASP), 229 [Google Scholar]
- Ehrenfreund, P., D’Hendecourt, L., Verstraete, L., et al. 1992, A&A, 259, 257 [NASA ADS] [Google Scholar]
- Fuchs, G. W., Cuppen, H. M., Ioppolo, S., et al. 2009, A&A, 505, 629 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Galvez, O., Belen, M., Herrero, V. J., & Escribiano, R. 2010, ApJ, 724, 539 [NASA ADS] [CrossRef] [Google Scholar]
- Gillis, H. A., & Quickenden, T. I. 2001, Canadian J. Chem. 79, 80 [Google Scholar]
- Gudipati, M. S., & Allamandola, L. J. 2003, ApJ, 596, L195 [NASA ADS] [CrossRef] [Google Scholar]
- Gudipati, M. S., & Allamandola, L. J. 2006, ApJ, 638, 286 [NASA ADS] [CrossRef] [Google Scholar]
- Guennoun, Z., Aupetit, C., & Mascetti, J. 2011, J. Phys. Chem. A, 115, 1844 [CrossRef] [Google Scholar]
- Hawkins, S., Kumi, G., Malyk, S., Reisler, H., & Wittig, C. 2005, Chem. Phys. Lett. 404, 19 [Google Scholar]
- Hudgins, D. M., & Allamandola, L. J. 1995, J. Phys. Chem., 99, 3033 [CrossRef] [PubMed] [Google Scholar]
- Hudgins, D. M., & Allamandola, L. J. 1997, J. Phys. Chem. A, 101, 3472 [CrossRef] [PubMed] [Google Scholar]
- Kamp, I. 2011, EAS Publ. Ser., 46, 271 [CrossRef] [EDP Sciences] [Google Scholar]
- Keane, J. V., Tielens, A. G. G. M., Boogert, A. C. A., Schutte, W. A., & Whittet, D. C. B. 2001, A&A, 376, 254 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Khan, A. 2004, J. Chem. Phys., 121, 280 [NASA ADS] [CrossRef] [Google Scholar]
- Kjaergaard, H. G., Robinson, T. W., & Brooking, K. A. 2000, J. Phys. Chem. A, 104, 11297 [CrossRef] [Google Scholar]
- Kumi, G., Malyk, S., Hawkins, S., Reisler, H., & Wittig, C. 2006, J. Phys. Chem. A, 110, 2097 [CrossRef] [Google Scholar]
- Malyk, S., Kumi, G., Reisler, H., & Wittig, C. 2007, J. Phys. Chem. A, 111, 13365 [CrossRef] [Google Scholar]
- Marsalek, O., Elles, C. G., Pieniazek, P. A., et al. 2011, J. Chem. Phys., 135, 224510 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Muñoz Caro, G. M., Meierhenrich, U. J., Schutte, W. A., et al. 2002, Nature, 416, 403 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Novakovskaya, Y. V., & Stepanov, N. F. 2004, Structural Chem., 15, 65 [CrossRef] [Google Scholar]
- Novozamsky, J. H., Schutte, W. A., & Keane, J. V. 2001, A&A, 379, 588 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Öberg, K. I., Garrod, R. T., van Dishoeck, E. F., & Linnartz, H. 2009, A&A, 504, 891 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Öberg, K. I., van Dishoeck, E. F., Linnartz, H, & Andersson, S. 2010, ApJ, 718, 832 [NASA ADS] [CrossRef] [Google Scholar]
- Park, J.-Y., & Woon, D. E. 2004a, ApJ, 601, L63 [NASA ADS] [CrossRef] [Google Scholar]
- Park, J.-Y., & Woon, D. E. 2004b, J. Phys. Chem A, 108, 6589 [CrossRef] [Google Scholar]
- Pontoppidan, K. M. 2004, Ph.D. Thesis, Leiden university [Google Scholar]
- Palumbo, M. E. 2006, A&A, 453, 923 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Romanescu, C., Marschall, J., Kim, D., Khatiwada, A., & Kalogerakis, K. S. 2010, Icarus, 205, 695 [Google Scholar]
- Roser, J. E., & Allamandola, L. J. 2010, ApJ, 722, 1932 [NASA ADS] [CrossRef] [Google Scholar]
- Salama, F., & Allamandola, L. J. 1991, J. Chem. phys., 94, 6964 [Google Scholar]
- Sandford, S. A., Bernstein, M. P., & Allamandola, L. J. 2004, ApJ, 607, 346 [NASA ADS] [CrossRef] [Google Scholar]
- Sceats, M. G., & Rice, S. A. 1983, Water: A Comprehensive Treatise, 7, eds. F. Franks (New York: Plenum) [Google Scholar]
- Schutte, W. A., & Greenberg, J. M. 1997, A&A, 317, L43 [NASA ADS] [Google Scholar]
- Schutte, W. A., & Khanna, R. K. 2003, A&A, 398, 1049 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Sellgren, K., Brooke, T. Y., Smith, R. G., & Geballe, T. R. 1995, ApJ, 449, L69 [NASA ADS] [CrossRef] [Google Scholar]
- Shida, T. 1988, Electronic Absorption Spectra of Radical Ions (Amsterdam: Elsevier) [Google Scholar]
- Steglich, M., Bouwman, J., Huisken, F., & Henning, Th. 2011, ApJ, 742, 2 [NASA ADS] [CrossRef] [Google Scholar]
- Steglich, M., Carpentier, Y., Jäger, C., et al. 2012, A&A, 540, A110 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Tielens, A. G. G. M. 2008, Annu. Rev. Astron. Astrophys., 46, 289 [Google Scholar]
- Van Dishoeck, E. F. 2006, IAU Symp. 251, eds. S. Kwok, & S. Sandford (Cambridge University Press) [Google Scholar]
- van Dishoeck, E. F., Jonkheid, B., & van Hemert, M. 2006, Faraday Disc., 133, 231 [Google Scholar]
- van Thiel, M., Becker, E. D., & Pimentel, G. C. 1957, J. Chem. phys., 27, 486 [Google Scholar]
- Verlet, J. R. R., Bragg, A. E., Kammrath, A., Cheshnovsky, O., & Neumark, D. M. 2005, Science, 307, 93 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Woon, D. E. 2004, Adv. Space Res., 33, 44 [NASA ADS] [CrossRef] [Google Scholar]
- Woon, D. E. 2006, AIP Conf. Proc. 855, eds. R. I. Kaiser, P. Bernath, A. M. Mebel, Y. Osamura, & S. Petrie (New York: Springer), 305 [Google Scholar]
- Woon, D. E., & Park, J.-Y. 2004, ApJ, 607, 342 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
Different PAH:H2O ice concentrations and temperatures studied here and the corresponding PAH photoionization yields.
Oscillator strengths and transition assignments of the neutral and cation transitions of coronene and pyrene considered here.
All Figures
|
Fig. 1 The 270−360 nm absorbance spectra of Cor:H2O ices at 5 different concentrations. All ices were deposited at 40 K. To aid comparison, all of the spectra are normalized to the peak of the S0–S3 transition. |
| In the text | |
|
Fig. 2 Absorbance spectrum (I) of coronene in H2O (1:14 000) at 12 K after ~12 min of vacuum UV photolysis referenced to the unphotolyzed spectrum (I0), shown at the bottom of Fig. 1. Negative peaks indicate the loss of neutral coronene while positive peaks indicate the appearance of new photoproducts, in this case the coronene cation. |
| In the text | |
|
Fig. 3 Growth of Cor+ and loss of Cor at different concentrations and different temperatures, as function of VUV dose (photolysis time). |
| In the text | |
|
Fig. 4 The 250−350 nm spectra of Py:H2O ices at 5 different concentrations. All ices were deposited at 40 K. |
| In the text | |
|
Fig. 5 Growth of Py+ and loss of Py at different concentrations and different temperatures, as function of VUV dose (photolysis time). |
| In the text | |
|
Fig. 6 Maximum ionization yield for different temperatures and concentrations of pyrene and coronene in H2O matrix. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.