| Issue |
A&A
Volume 544, August 2012
|
|
|---|---|---|
| Article Number | A82 | |
| Number of page(s) | 5 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/201219594 | |
| Published online | 01 August 2012 | |
The submillimeter-wave spectrum of diisocyanomethane⋆
1 Laboratoire de Physique des Lasers, Atomes, et Molécules, UMR CNRS 8523, Université de Lille 1, 59655 Villeneuve d’Ascq Cedex, France
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2 Sciences Chimiques de Rennes, UMR 6226 CNRS – ENSCR, Avenue du Général Leclerc, CS 50837, 35708 Rennes Cedex 7, France
Received: 14 May 2012
Accepted: 28 June 2012
Abstract
Context. Several organic isocyanides (HNC, CH3NC, HCCNC) have been observed to date in the interstellar medium (ISM). However there is still a lack of high-resolution spectroscopic data for simple isocyanides that could provide a basis for their detection.
Aims. Diisocyanomethane (CNCH2NC) is a small molecule with only seven atoms that is an interesting candidate for astrophysical detection. The rotational spectrum of diisocyanomethane has never been studied before, hence we present our analysis of its spectrum.
Methods. We measured the rotational spectrum of diisocyanomethane in the frequency range 120−620 GHz using the Lille fast-scan and solid-state source spectrometers. The spectroscopic study was supported by high-level theoretical calculations of the molecular structure and both the harmonic and anharmonic force field.
Results. The ground and the first excited vibrational state (ν15) are assigned and analysed. The dataset is composed of more than 2000 measured and fitted lines, which allows us to make accurate predictions of transition frequencies of diisocyanomethane in the frequency range up to 900 GHz. The statistical analysis of the results of the fit shows that the use of the S-reduction of the Watson rovibrational Hamiltonian for spectral modelling is more appropriate.
Key words: ISM: molecules / methods: laboratory / submillimeter: ISM / molecular data / line: identification
Full Tables 2 and 3 are only available at the CDS via anonymous ftp to cdsarc.u-strasbg.fr (130.79.128.5) or via http://cdsarc.u-strasbg.fr/viz-bin/qcat?J/A+A/544/A82
© ESO, 2012
1. Introduction
In the interstellar medium (ISM), about 160 compounds have been detected to date (see, for example, Guillemin 2011). Carbon, hydrogen, nitrogen, and oxygen are the main represented elements. Most species have a small number of atoms (fewer than nine for 90% of them); cyanides and carbonyl derivatives are particularly abundant. About 20% of these compounds possess two heteroatoms, but surprisingly no dinitrile has so far been detected. In the case of polyynes, the presence of a nitrile group at an extremity of the molecule leads to a compound with a high dipole moment leading to an easily detectable species, and the cyanopolyynes constitute the most abundant family of compounds detected in this medium. The lack of dicyanopolyynes and the few polyynes observed in the ISM can be easily explained even if these derivatives could be abundant in this medium: for ethyne or polyynes with a nitrile group or hydrogen at each extremity, the permanent dipole moment is 0 and they cannot be detected by microwave spectroscopy but only by infrared spectroscopy.
Isocyanides are isomers of the corresponding nitriles and some have already been observed in the ISM (HNC, CH3NC, HCCNC). Looking for small molecules with two nitrogen atoms and a significant dipole moment, we investigated some synthetic and spectroscopic studies on such compounds. As a tool promoting its detection in the ISM if it were present there, we report here the microwave spectrum of the diisocyanomethane (CNCH2NC), which is a quite small molecule with only seven atoms and two nitrogen atoms. The studied compound is an isomer of the more stable malonitrile (NCCH2CN). In general, the spectroscopic studies of isocyanic molecules are rather sparse compared to their cyanic isomers. This may arise from difficulties in the synthesis associated with the foul odour of these compounds and the kinetic instability of many of them leading to additional problems in experimental spectroscopy. Although several microwave studies have been recently performed on isocyanides (Møllendal et al. 2011a,b; Samdal et al. 2011), this work is probably the first microwave study of a bis-isocyanide derivative.
2. Experiments
The synthesis of diisocyanomethane was reported by Neidlein but its presence was only proven by derivatization (Neidlein 1964). Fifteen years ago, this compound was easily prepared by a bis-dehydration of the corresponding bis-formamide and unambiguously isolated (Buschmann et al. 1997). A detailed preparation was then reported (Bartolomäs et al. 2002). The white solid obtained at low temperature (−40 °C) can explode on heating (when its temperature exceeds −10 °C), but some spectra were recorded: nuclear magnetic resonance (NMR), Raman, and infrared (IR) spectra assisted by theoretical calculations and the X-ray crystal structure of a cooled sample (Bartolomäs et al. 2002). Using the same approach, we prepared samples of about a half-gram and the trap used to selectively condense the product during the purification step was directly connected to the absorption cell of the spectrometer.
The first measurements of the rotational spectrum of diisocyanomethane were performed in the frequency range 120−180 GHz using the Lille fast-scan spectrometer (Alekseev et al. 2012). The use of a fast-scan technique can be very important at the initial stage of studies of unstable species. While the stability of the sample in the absorption cell is rather questionable, the fast scan technique provides a rapid acquisition of broadband spectra and allows the optimization of the experimental conditions in a minimum amount of time. Two different absorption cells were used during the measurements. At first, spectra were recorded using a stainless-steel absorption cell that was 6 cm in diameter and 2.1 m long. These spectra contained few lines with low signal-to-noise ratio. Significantly better results were obtained by the use of a 1.4 m long Pyrex cell. In this case, the recorded spectra were much more intense and easily allowed their assignment. Thus, one can conclude that metallic surface accelerates decomposition of the studied compound. Both absorption cells were used in the so-called “flow mode”, i.e. the sample of diisocyanomethane was evaporated outside the cell and injected into it through a side opening at one end of the cell and continuously pumped out through another side opening at the other end. The optimal temperature of evaporation was found to be close to −25 °C.
The second series of measurements were performed in the range 180−620 GHz using the sub-mm wave spectrometer based on a frequency multiplication chain (Motiyenko et al. 2010). Owing to the limited amount of sample and lower acquisition rate (compared to fast-scan spectrometer), only the regions of approximately 10 GHz wide were scanned in the vicinity of 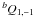 bands. The measurement accuracy of a strong isolated line is estimated to be 0.02 MHz for fast-scan spectra, 0.03 MHz for spectra recorded in the frequency range up to 500 GHz, and 0.05 MHz for spectra in the frequency range above 500 GHz.
bands. The measurement accuracy of a strong isolated line is estimated to be 0.02 MHz for fast-scan spectra, 0.03 MHz for spectra recorded in the frequency range up to 500 GHz, and 0.05 MHz for spectra in the frequency range above 500 GHz.
3. Quantum chemical calculations
The spectroscopic work was preceded by quantum chemical calculations in order to provide an initial basis for spectral assignments. The ab initio and density functional theory (DFT) calculations were performed by employing the Gaussian 09 suite of programs (Frisch et al. 2009). Calculations were performed using Møller-Plesset second-order perturbation calculations (MP2) (Møller & Plesset 1934), and density functional theory (DFT) calculations employing Beckes three-parameter hybrid functional (Becke 1988) and the Lee, Yang and Parr correlation functional (B3LYP) (Lee et al. 1988). The Peterson and Dunning’s (Peterson & Dunning 2002) correlation-consistent triple-ζ wave function augmented with the diffuse function aug-cc-pVTZ was employed in the MP2 calculations. The 6-311++G(3df, 3pd) wave function augmented with diffuse functions, was employed in the B3LYP calculations.
The results of our calculations include structural optimization as well as harmonic force field parameters in order to provide the information about the rotational and quartic centrifugal distortion constants, as well as the low frequency vibrational modes. In addition, cubic force field calculations were performed at the B3LYP level in order to estimate the values of the sextic centrifugal distortion constants. These results are given in Tables A.1–A.3.
Rotational constants of diisocyanomethane.
4. Assignments and fit
Diisocyanomethane is a prolate asymmetric top molecule not too far from a symmetric top limit (Ray’s asymmetry parameter, κ = −0.96). In this case, the Watson’s rovibrational Hamiltonian (Watson 1977) is usually set up in the Ir coordinate representation, although the choice of either A-reduced or S-reduced form of the Hamiltonian is not obvious. These two reductions imply different definitions of s111 parameter of the transformation (reduction) operator. One of the first criteria to be used in order to choose a reduction is the value of s111, which should be as small as possible. The value of s111 parameter was estimated from the ab initio harmonic force field. It appears that for both reductions of the Hamiltonian, the values of s111 are rather close (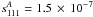 and
and 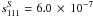 ) thus making it difficult to use this criterion in order to choose between A- and S-reduction. Therefore, in the analysis of the spectrum we used both reductions of the Hamiltonian, and the final choice was based on other criteria such as the residuals of the fit, its standard deviation, the number of fitted parameters, and the condition number of the system of normal equations (Margulès et al. 2010).
) thus making it difficult to use this criterion in order to choose between A- and S-reduction. Therefore, in the analysis of the spectrum we used both reductions of the Hamiltonian, and the final choice was based on other criteria such as the residuals of the fit, its standard deviation, the number of fitted parameters, and the condition number of the system of normal equations (Margulès et al. 2010).
From the results of the ab initio calculations, it follows that the molecular dipole moment of diisocyanomethane is close to 3.5 D and coincides with the intramolecular axis b. Thus, only b-type transitions were expected in the spectra. At the initial stage of assignment, we searched for pairs of the strongest 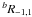 and
and 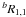 transitions with Ka = 0 and 1. They were found relatively close (within a few tens of MHz) to the positions predicted on the basis of rotational and quartic centrifugal distortion constants from ab initio calculations. The following assignment was performed in a classical bootstrap manner, where assigned transitions were used to improve the frequency predictions and search for new ones. The final dataset consists of 1345 lines corresponding to 1919 rotational transitions. The maximum values of the J and Ka quantum numbers for the transitions used in the fit are, respectively, 99 and 17.
transitions with Ka = 0 and 1. They were found relatively close (within a few tens of MHz) to the positions predicted on the basis of rotational and quartic centrifugal distortion constants from ab initio calculations. The following assignment was performed in a classical bootstrap manner, where assigned transitions were used to improve the frequency predictions and search for new ones. The final dataset consists of 1345 lines corresponding to 1919 rotational transitions. The maximum values of the J and Ka quantum numbers for the transitions used in the fit are, respectively, 99 and 17.
In addition to the ground vibrational state, the first excited state ν15 of the mode corresponding to symmetrical out-of-plane vibrations of the two NC groups was assigned in the spectra recorded. According to an the ab initio calculation, the ν15 state lies 127 cm-1 above the ground state and its Boltzmann factor at room temperature is 0.6. The rotational transitions of the ν15 state were assigned in a manner similar to the ground state, starting with vibrational satellites of the and lines with Ka = 0 and 1. The assignment was also facilitated by the use of the centrifugal distortion constants of the ground state as a basis for the initial frequency predictions. The final dataset for the ν15 state consists of 873 lines having maximum values of J and Ka of, respectively, 85 and 17.
A part of the table available at CDS, with assigned rotational transitions of the ground state of diiscocyanomethane.
 |
Fig. 1 A portion of the rotational spectrum of diisocyanomethane illustrating the influence of nuclear spin statistics on transition intensities. In red: experimental spectrum; in blue: two theoretical spectra calculated in assumption of equivalent statistical weights and in assumption of even/odd = 15/21. |
A part of the table available at CDS, with assigned rotational transitions of the ν15 = 1 excited vibrational state of diiscocyanomethane.
The two datasets of the ground and ν15 states were fitted using A- and S-reductions of the Watson’s Hamiltonian in Ir coordinate representation and the results of the fits are given in Table 1. As follows from the comparison of the results presented in Table 1, the performance of both methods is rather similar. At first look, the use of the A-reduction seems to be more efficient in terms of rms and weighted rms deviation. However, the condition number of the fit (Margulès et al. 2010) of the ground state is too big and indicates that there is a rather poor conditioning of the system of normal equations in the case of the A-reduction. A thorough inspection of correlation matrix has indeed revealed an unusual and rather strong correlation between the octic parameters LJK and lK. Fixing one of them to 0 led to a fit with a condition number similar to the value obtained using the S-reduction, but with a much higher rms deviation (about 0.06 MHz). It appears that using the A-reduction, one cannot fit all the ground state transition frequencies within the experimental accuracy by omitting either the LJK or lK parameter. In the final fit, both parameters were retained despite of rather bad conditioning. The condition number in the case of the S-reduction is much smaller. However, 20 lines were excluded from the final fit using the S-reduction as their frequency residuals did not satisfy a 3σ criterion, where σ is the experimental measurement uncertainty. All of them belong to a series of high-J (J > 80) and low-Ka transitions. For the first excited state ν15, the conditioning is reasonable for the fit using A-reduction as there is no any strong correlation between parameters of the same order. And the condition number is remarkably small for the case of S-reduction, while five transitions of the ν15 state with frequency residuals exceeding 3σ were excluded from the fit. The complete list of measured rotational transitions of the ground and ν15 states is presented in Tables 2, 3 available at the CDS1. Here, only parts of Tables 2 and 3 are shown as examples.
One should also note that the intensities of rotational transitions of diisocyanomethane are subjected to nuclear spin statistics. The rotation about the b axis by π radians, or, equivalently, the C2b operation of the C2v symmetry point group, simultaneously exchanges the positions of two nitrogen and two hydrogen atoms in the molecule. Since the nitrogen nuclei are bosons (I1 = 1) and the hydrogen nuclei are fermions (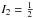 ), the resultant statistics is Fermi-Dirac. Therefore, the total wavefunction ψtot = ψeψvψrψn must be antisymmetric with respect to the C2b operation. For the ground electronic and vibrational states, the corresponding wavefunctions ψe and ψv are symmetric. The parity of the rotational wavefunction ψr depends on the Ka and Kc values and for the levels with Ka + Kc = evenψr is symmetric, while ψr for the levels with Ka + Kc = odd is antisymmetric. Finally, a symmetric nuclear spin function must be combined with an antisymmetric rotational wavefunction and an antisymmetric nuclear spin function must be combined with a symmetric rotational wavefunction in order to satisfy Fermi-Dirac statistics. The total number of nuclear spin function is (2I1 + 1)2(2I2 + 1)2 = 36. Taking into account the nuclear spin statistics for the ground vibrational state of diisocyanomethane, the nuclear statistical weight is 15 for the rotational levels with Ka + Kc = even, and 21 for the rotational levels with Ka + Kc = odd. The influence of nuclear spin statistics on the intensities of rotational lines is illustrated in Fig. 1, where experimental intensities of a series of transitions of the ground state are compared to theoretical predictions calculated with statistical weights of 1/1 and 15/21.
), the resultant statistics is Fermi-Dirac. Therefore, the total wavefunction ψtot = ψeψvψrψn must be antisymmetric with respect to the C2b operation. For the ground electronic and vibrational states, the corresponding wavefunctions ψe and ψv are symmetric. The parity of the rotational wavefunction ψr depends on the Ka and Kc values and for the levels with Ka + Kc = evenψr is symmetric, while ψr for the levels with Ka + Kc = odd is antisymmetric. Finally, a symmetric nuclear spin function must be combined with an antisymmetric rotational wavefunction and an antisymmetric nuclear spin function must be combined with a symmetric rotational wavefunction in order to satisfy Fermi-Dirac statistics. The total number of nuclear spin function is (2I1 + 1)2(2I2 + 1)2 = 36. Taking into account the nuclear spin statistics for the ground vibrational state of diisocyanomethane, the nuclear statistical weight is 15 for the rotational levels with Ka + Kc = even, and 21 for the rotational levels with Ka + Kc = odd. The influence of nuclear spin statistics on the intensities of rotational lines is illustrated in Fig. 1, where experimental intensities of a series of transitions of the ground state are compared to theoretical predictions calculated with statistical weights of 1/1 and 15/21.
5. Conclusions
The spectroscopic information obtained in the present study should be fairly accurate enough to provide the basis for the astrophysical detection of diisocyanomethane. The present analysis covers the frequency range where the most intense transitions are expected at low temperatures. It allows us to obtain accurate frequency predictions at least for transitions involving levels with J ≤ 80 and Ka ≤ 17 and in the frequency range up to 900 GHz. The predictions should be also accurate in terms of their extrapolation to a lower frequency range where there may be a greater chance of an astronomical detection owing to the smaller amount of line crowding. For this purpose, the use of the S-reduction seems to be more appropriate as it is the one that provides a better conditioning of the fit. Owing to convergence problems and ill-conditioning, the transitions having higher values of J and Ka could be predicted with significant errors. A possible explanation of this phenomenon is the existence of Fermi-type local interactions between the ground and ν15 state. There are no symmetry-allowed Coriolis terms that can connect the two states, since both of them belong to the A1 irreducible representation of the C2v symmetry group. The interactions could be much better accounted for using operators of the A-reduced Hamiltonian, but at the same time this could also lead to additional correlation problems. At the same time, the levels with J and Ka that exceed the aforementioned values are poorly populated especially at low temperatures and, thus, would not be of any interest to the compilation of astrophysical databases.
Acknowledgments
This work was supported by the French program Action sur Projets de l’INSU “Physique et Chimie du Milieu Interstellaire”. J.-C.G. thanks the Centre National d’Études Spatiales (CNES) for Financial support.
References
- Alekseev, E. A., Motiyenko, R. A., & Margulès, L. 2012, Radio Physics and Radio Astronomy, 3, 75 [NASA ADS] [CrossRef] [Google Scholar]
- Bartolomäs, T., Lentz, D., Neubert, I., & Röttger, M. 2002, Anorg. Allg. Chem., 628, 863 [CrossRef] [Google Scholar]
- Becke, A. D. 1988, Phys. Rev. A, 38, 3098 [Google Scholar]
- Buschmann, J., Bartolmas, T., Lentz, D., et al. 1997, Angew. Chem. lnt. Ed. Engl., 36, 2372 [CrossRef] [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. 2009, Gaussian 09, revision A.1, Gaussian, Inc., Pittsburgh PA [Google Scholar]
- Guillemin, J.-C. 2011, EpJ Web Conf., 18, 06004 [CrossRef] [EDP Sciences] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Margulès, L., Motiyenko, R. A., Alekseev, E. A., & Demaison, J. 2010, J. Mol. Spectrosc., 260, 23 [Google Scholar]
- Møllendal, H., Samdal, S., Matrane, A., & Guillemin, J.-C. 2011a, J. Phys. Chem. A, 115, 7978 [CrossRef] [Google Scholar]
- Møllendal, H., Samdal, S., & Guillemin, J.-C. 2011b, J. Phys. Chem. A, 115, 13474 [CrossRef] [Google Scholar]
- Møller, C., & Plesset, M. S. 1934, Phys. Rev., 46, 618 [NASA ADS] [CrossRef] [Google Scholar]
- Motiyenko, R. A., Margulès, L., Alekseev, E. A., Guillemin, J. C., & Demaison, J. 2010, J. Mol. Spectrosc., 264, 94 [Google Scholar]
- Neidlein, R. 1964, Angew. Chem. Int. Ed. Engl., 3, 382 [CrossRef] [Google Scholar]
- Peterson, K. A., & Dunning, T. H., Jr. 2002, J. Chem. Phys., 117, 10548 [NASA ADS] [CrossRef] [Google Scholar]
- Samdal, S., Mollendal, H., & Guillemin, J.-C. 2011, J. Phys. Chem. A, 115, 9192 [CrossRef] [Google Scholar]
- Watson, J. K. G. 1977, Vibrational Spectra and Structure (Amsterdam: Elsevier) [Google Scholar]
Appendix A: The results of ab initio calculations on diisocyanomethane
The calculated MP2/aug-cc-pVTZ molecular structure of diisocyanomethane.
The MP2/aug-cc-pVTZ and B3LYP/6-311G(3df,2pd) spectroscopic parameters of diisocyanomethane.
| A-reduction | S-reduction | ||
| Parameter | Value | Parameter | Value |
|
|
|||
| MP2/aug-cc-pVTZ | |||
| A (MHz) | 21 877.85 | A(MHz) | 21 877.85 |
| B (MHz) | 3329.57 | B(MHz) | 3329.56 |
| C (MHz) | 2943.33 | C(MHz) | 2943.35 |
| ΔJ (kHz) | 2.494 | DJ(kHz) | 2.451 |
| ΔJK (kHz) | –92.06 | DJK(kHz) | –91.81 |
| ΔK (kHz) | 1171.7 | DK(kHz) | 1171.5 |
| δJ (kHz) | 0.725 | d1(kHz) | –0.725 |
| δK (kHz) | 8.304 | d2(kHz) | –0.0213 |
| B3LYP/6-311G(3df, 2pd) | |||
| ΦJ (Hz) | 0.0108 | HJ(Hz) | 0.0104 |
| ΦJK (Hz) | –0.264 | HJK(Hz) | –0.415 |
| ΦKJ (Hz) | –2.969 | HKJ(Hz) | –2.463 |
| ΦK (Hz) | 91.54 | HK(Hz) | 91.19 |
| φJ (Hz) | 0.0044 | h1(Hz) | 0.0043 |
| φJK (Hz) | –0.0252 | h2(Hz) | 1.67 × 10-3 |
| φK (Hz) | 11.32 | h3(Hz) | 9.04 × 10-5 |
The MP2/aug-cc-pVTZ harmonic vibrational frequencies of diisocyanomethane.
All Tables
A part of the table available at CDS, with assigned rotational transitions of the ground state of diiscocyanomethane.
A part of the table available at CDS, with assigned rotational transitions of the ν15 = 1 excited vibrational state of diiscocyanomethane.
All Figures
|
Fig. 1 A portion of the rotational spectrum of diisocyanomethane illustrating the influence of nuclear spin statistics on transition intensities. In red: experimental spectrum; in blue: two theoretical spectra calculated in assumption of equivalent statistical weights and in assumption of even/odd = 15/21. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.