| Issue |
A&A
Volume 535, November 2011
|
|
|---|---|---|
| Article Number | A74 | |
| Number of page(s) | 12 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201117103 | |
| Published online | 11 November 2011 | |
Initiating molecular growth in the interstellar medium via dimeric complexes of observed ions and molecules⋆
1
MS: 245-6, Space Science and Astrobiology Division, NASA Ames Research Center, Mountain View, 94035 California, USA
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
2
Department of Chemistry, University of California and Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, 94720 California, USA
3
MS: 245-1, Space Science and Astrobiology Division, NASA Ames Research Center, Mountain View, 94035 California, USA
Received: 19 April 2011
Accepted: 18 July 2011
Abstract
A feasible initiation step for particle growth in the interstellar medium (ISM) is simulated by means of ab initio quantum chemistry methods. The systems studied are dimer ions formed by pairing nitrogen containing small molecules known to exist in the ISM with ions of unsaturated hydrocarbons or vice versa. Complexation energies, structures of ensuing complexes and electronic excitation spectra of the encounter complexes are estimated using various quantum chemistry methods. Møller-Plesset perturbation theory (MP2), Z-averaged perturbation theory (ZAPT2), coupled cluster singles and doubles with perturbative triples corrections (CCSD(T)), and density functional theory (DFT) methods (B3LYP) were employed along with the correlation consistent cc-pVTZ and aug-cc-pVTZ basis sets. Two types of complexes are predicted. One type of complex has electrostatic binding with moderate (7−20 kcal mol-1) binding energies, that are nonetheless significantly stronger than typical van der Waals interactions between molecules of this size. The other type of complex develops strong covalent bonds between the fragments. Cyclic isomers of the nitrogen containing complexes are produced very easily by ion-molecule reactions. Some of these complexes show intense ultraviolet-visible spectra for electronic transitions with large oscillator strengths at the B3LYP, ωB97, and equations of motion coupled cluster (EOM-CCSD) levels. The open shell nitrogen containing carbonaceous complexes especially exhibit a large oscillator strength electronic transition in the visible region of the electromagnetic spectrum.
Key words: astrochemistry / acceleration of particles / molecular processes
Appendix A is available in electronic form at http://www.aanda.org
Corresponding author email: This email address is being protected from spambots. You need JavaScript enabled to view it.
© ESO, 2011
1. Introduction
In the interstellar medium (ISM) thus far, inorganic salts, neutral hydrocarbons, olefins, cumulenes, nitriles, alcohols, carboxylic acids, cations, and anions have been identified (Gerakins 2011). Some of these are in the gas phase, and some are in interstellar ices and grains. Large polycyclic aromatic hydrocarbons (PAH) are also known to exist in the ISM, as a general class of interstellar molecules (Chillier et al. 1999; Salama et al. 1999). Recently, two fullerene molecules (C60 and C70) have been identified in a carbon rich planetary nebulae (Cami et al. 2010). It is proposed that the identification of individual PAH molecules could help to resolve some of the mysteries associated with the diffuse interstellar bands (DIBs; Salama et al. 1999; Snow 2001) and the galactic extended red emission (ERE; Rhee et al. 2007). Similar to PAHs, spectroscopic signatures of nitrogen containing polycyclic aromatic hydrocarbons (PANH) are prevalent in various cosmological environments (Hudgins et al. 2005). Nitrogen containing molecules and ions have been observed throughout the ISM. Gas phase reactions of nitrogen containing species with carbonaceous compounds represent an ideal way to incorporate nitrogen into carbon chain growth, and ultimately into cyclic and polycyclic carbon compounds. Although the smaller molecules are pervasive in the ISM, how large molecules, chains, polycyclic complexes, and grains are formed in the ISM remains an open question.
Weak intermolecular interactions dipole-dipole, ion-induced dipole, hydrogen bonding, and dispersion (van der Waals) forces between atoms, molecules, and ions, play major roles in nature. For example, in biological systems hydrogen bonding helps to keep a whole DNA strand together. In the gas phase, long-range ion-dipole and electrostatic interactions guide species to come together to form aggregates. In the very low particle density cold ISM, in the absence of other strong forces, these weak interactions can guide ions and molecules to condense into aggregates and initiate reactions (Herbst 2001; Smith 2002). Ions are relatively abundant species in the strong radiation fields of the ISM, in star forming regions, in planetary nebulae, and in boundary regions between hot and cold environments. Ion-molecule reactions are barrierless and spontaneous in the gas phase. It is reasonable, therefore, to ask if this cold condensation can be a viable mechanism for forming small molecular complexes, large PAHs, and grains? And, if so, what are the specific pathways?
Earlier theoretical and experimental studies have concentrated on the π-π interactions leading to stable complexes in alkenes (Bates et al. 2008; Hopkins & Tschumper 2004), aromatic systems (Sinnokrot & Sherrill 2004), and even in PAHs (Hirata et al. 1999; Szczepanski & Vala 1993; Weisman et al. 2001; Weisman et al. 2005). The neutral complexes are typically bound by van der Waals interactions (though aromatic radical-radical stacking complexes with partial covalent character are known (e.g., Small et al. 2005). However, the cationic complexes are bound by both electrostatic and covalent interactions. Closed and open shell PAH dimers are estimated to be bound by energies of about 20 kcal mol-1, and absorb in the visible and sometimes in the infrared (Rhee et al. 2007; Weisman et al. 2005). The cationic complexes of small olefins, especially the nitrogen containing ones, have not been explored in any detail.
The objective of the current study is threefold. Firstly, we have examined, by means of ab initio quantum chemical methods, the nature of binding among the simplest complexes between some observed interstellar molecules and ions. In other words, the question of how the small precursors complex together to form larger aggregates in interstellar low-density, low temperature conditions is explored. Secondly, we have computed the UV-Vis electronic spectra and the vibrational spectra of these species so that they may be compared with experiments as well as existing astronomical observations. It is vital to know the structural and spectroscopic nature of the ensuing complexes to understand their formation pathways. Some of these ion complexes may have large oscillator strengths in their electronic spectra due to charge transfer excitations. Thirdly, we have performed a systematic study of the complexes using various ab initio quantum chemistry methods. Binding energies, excitation energies and geometrical parameters obtained using affordable density functional and perturbation theory methods were compared and benchmarked with results obtained by highly accurate coupled cluster methods. This benchmarking study will help us choose more affordable ab initio quantum chemistry methods for charting out pathways of formation of larger molecules in the future where coupled cluster calculations may not be computationally feasible.
Several studies have examined the radiative association reaction rates for relatively simple cation/neutral reactions such as C+ + H2, and even up to systems with three heavy (non-hydrogen) atoms under ISM conditions (Black et al. 1975; Smith 1992; Gerlich & Horning 1992; Barinovs & van Hemert 2006; Herbst et al. 2010). While our study is related to these investigations, examining formation products and their spectroscopic signatures, we have not investigated the radiative association rates, though we note that the studies cited above have demonstrated that in general radiative association rates increase as the cations/neutrals increase in size, which of course is consistent with the larger number of rovibrational degrees of freedom.
Binding energies (in kcal mol-1) of the dimer cations between (mostly) known interstellar molecules and cations are listed.
Binding energies (in kcal mol-1) of the open shell complexes of astrochemically observed species.
2. Theoretical methods
The geometrical structures reported in this research are optimized using second order Møller-Plesset perturbation theory (MP2) along with Dunnings correlation consistent polarized valence triple zeta basis set (cc-pVTZ) and the augmented correlation consistent polarized valence triple zeta basis set (aug-cc-pVTZ; Dunning 1989). MP2/aug-cc-pVTZ optimized geometries were used when calculating the binding energies of the complexes employing the coupled cluster singles and doubles with a perturbative triples correction, CCSD(T), method (Purvis & Bartlett 1982; Raghavachari et al. 1989; Lee & Scuseria 1995), in conjunction with the cc-pVTZ and aug-cc-pVTZ basis sets. The binding energies were calculated by subtracting the absolute energies of the neutral and the cationic fragments from the energy of the cationic bound complex. Binding energies were also determined using a density functional theory (DFT) method for comparison. We employ Becke’s widely used three-parameter hybrid functional (Becke 1993), based on the Becke exchange functional (Becke 1988) paired up with the correlation functional of Lee et al. (LYP; Lee et al. 1988). All geometries were fully optimized using B3LYP along with the cc-pVTZ basis set. A fine grid of 75 radial points and 590 angular functions was used for all the DFT computations. Open shell molecules were treated with unrestricted MP2 (UMP2), second order Z-averaged perturbation theory (ZAPT2) (Lee & Jayatilaka 1993; Lee et al. 1994), and UB3LYP methods along with the cc-pVTZ basis set. The ZAPT2 method is based on the use of a symmetric spin orbital basis (Jayatilaka & Lee 1992), and leads to Sx eigenfunctions in the n-particle space. It has been shown to have less spin contamination relative to UMP2 and other restricted open-shell perturbation theories, and to have a larger radius of convergence, and to be less costly (computationally) as well (Lee et al. 1994). For electronic excitation energies, starting from the ground electronic state to various singlet excited states, including oscillator strengths, time dependent density functional theory (TDDFT) (Hirata et al. 1999) computations were carried out using the B3LYP density functional and the cc-pVTZ basis set. A range separated density functional, ωB97 (Chai & Head-Gordon 2008), that is specifically designed for weak interactions was also employed. For valence electronic transitions TDDFT has been shown to work well (Hirata et al. 1999; Weisman et al. 2001; Halasinski et al. 2003; Hirata et al. 2003). The range-separated functional, ωB97, significantly reduces the serious problems of TDDFT with conventional functionals for charge-transfer excited states (Dreuw & Head-Gordon 2004). The Q-Chem3.1 (Shao et al. 2006) ab initio quantum chemistry code was used for all the DFT, TDDFT, MP2, (and UMP2) and ZAPT2 computations. Coupled cluster calculations were carried out using the MOLPRO 2006 (Werner & Knowels 2006) quantum chemistry package. Harmonic vibrational frequency calculations were carried out at the MP2/cc-pVTZ and MP2/aug-cc-pVTZ levels to ensure that the molecules and complexes presented here represent true minima. The Cartesian coordinates of all the species discussed in this work are included in the supplementary material available with the web site.
3. Results
Elementary growth processes starting from some of the nitrogen containing molecules or ions observed in interstellar space are investigated in the following sections. Specifically, several nitrogen containing interstellar molecules and cations that have been observed in the ISM were paired up with small, unsaturated hydrocarbons to simulate the initial nucleation step that could then be a pathway to formation of larger molecules, such as PAHs and PANHs, and ultimately to grain formation. Cationic complex formation is guided by long-range weak electrostatic ion-dipole type attraction, in which a wide variety of exotic products are expected. Some ion-molecule reactions in the low temperature and ultra low-density conditions of the ISM readily make strong covalent bonds to produce linear or branched chains, replication of which leads to chain growth. In the next section we will see that these types of reactions take place with significant favorable energy changes for larger molecule formation. Other dimers make relatively weak and chemically interesting face-to-face π-stacked or T-shaped complexes bound by electrostatic or ion-dipole type interactions. Although several association or encounter complexes are investigated in depth for each system, only the simplest kinds of encounter complexes are included in this work as it is not intended to be an extensive search of the entire potential energy surfaces for all possible minima of the molecular complexes. That is, complex or molecule formation in which a significant potential barrier must be overcome are not investigated in the present study. Binding energies for closed-shell complexes are presented in Table 1 and similarly for open-shell complexes in Table 2. In Tables 3 and 4, the electronic excitation energies are presented at the B3LYP/cc-pVTZ and ωB97/cc-pVTZ levels of theories. In Table 3, the excitation energies of the complexes are given, while in Table 4 those of the monomeric molecules and ions are listed. While this paper is focused on nitrogen containing complexes, we have additionally studied the pure carbon complexes of HCCH, H2CCH2 with their cations, which will be presented in another paper (Bera et al., in prep.).
TDDFT electronic excitation energies (in eV) of the complexes calculated with B3LYP/cc-pVTZ and ωB97/cc-pVTZ.
3.1. Closed shell cation
I. [C2H4–HCNH]+.
Ethylene, hydrogen cyanide and protonated hydrogen cyanides are observed species in some carbonaceous interstellar clouds (Snyder & Buhl 1971; Ziurys & Turner 1986). Electrostatic interaction between protonated hydrogen cyanide and ethylene leads to two C2v symmetry stable T-shaped complexes and two cyclic isomers with favorable binding energies. The CH hydrogen in HCNH+ interacts with the π electron clouds of H2CCH2 to make the stable complex A presented in Fig. 1.A. The distance between the H atom and either of the C atoms of ethylene is 2.156, and the binding energy is 13.24 kcal mol-1 at the geometry optimized at the MP2/aug-cc-pVTZ level of theory. As shown in Table 1, the binding energy at the higher CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ level of theory is 11.7 kcal mol-1 (henceforth we will abbreviate this as the CCSD(T)/aug-cc-pVTZ binding energy). The NH hydrogen in HCNH+ also interacts favorably with ethylene to make a more stable isomer, B, of this complex, presented in Fig. 1.B. The NH–C distance is 1.912 at the MP2/aug-cc-pVTZ optimized geometry. The more tightly bound nature of the complex is reflected in the larger binding energy, 16.58 kcal mol-1 at the CCSD(T)/aug-cc-pVTZ level, as presented in Table 1.
Although, formation of the lower energy isomer is expected due to thermodynamic reasons, the branching ratio for formation of the two isomers will depend on the approach geometries of the two constituents, and their relative translational energy. The apparent branching ratio may also depend upon the barrier to internal conversion between the isomers.
The electronic excitation energies (in eV) and oscillator strengths are presented in Table 3 for all the complexes calculated using TDDFT as stated previously. Both have very strong oscillator strength absorption features in the UV. As seen from Table 3, B3LYP under predicts the excitation energies of these weakly bound complexes compared to the ωB97 functional, which reduces self-interaction errors. The ωB97 results (5.56 eV) are much closer to the excitation energies obtained at the EOM-CCSD/cc-pVTZ level of theory (6.46 eV) for the complex 1A. For complex 1B the EOM-CCSD excitation energy for the first excited state is 7.22 eV compared to 6.57 eV obtained with the ωB97 functional.
Two other isomers with more traditional covalent bonding features are also located on the potential energy surface of the association complex. These are a four-membered ring and a three-membered ring structure. The four-membered ring isomer presented in Fig. 1.C is bound by 50.07 kcal mol-1 energy at the CCSD(T)/aug-cc-pVTZ level, making it the most strongly bound of the isomers studied. By contrast, however, the three-membered ring structure (D) is the most weakly bound of the structures presented in Fig. 1.D, with only 1.82 kcal mol-1 of binding energy. A comparison of the association energies for the complexes using the B3LYP, MP2 and CCSD(T) methods is presented in Table 1. Both B3LYP and MP2 estimated association energies match very well in comparison to CCSD(T). The boat (Fig. 1.C) and the bridge (Fig. 1.D) isomers have intense electronic absorptions at 7.99 eV and 8.54 eV respectively.
Association energies for a complex between an ionic and a neutral species are generally large and thus are not going to be significantly changed by the basis set superposition error (BSSE) correction. One possible exception is the case of the bridge structure (Fig. 1.D) for which the association energy is small (1.96 kcal/mol). However, as Fig. 1.D shows, after association, the geometry changes significantly from the initial monomer geometries, which also explains why the association energy for an ion/neutral complex is small in this case in the association some new bonds are formed, but also some other bonds are weakened. A BSSE correction in the situation where the two monomer units do not retain their monomer quality but rather react to become one molecule is uninformative. Thus, BSSE corrections have not been computed for the ion/neutral complexes included in the present study.
Excitation energies (in eV) and oscillator strengths of selected molecules and ions using time-dependent density functional theory (TDDFT) with B3LYP/cc-pVTZ and ωB97/cc-pVTZ.
II. [H2CCH2–H2CNH2] + .
Methyleneimine, H2CNH, is observed in various interstellar environments (Johnson & Lovas 1972). Planar protonated methyleneimine can bind with ethylene by a face-to-face stacking interaction. The parallel-displaced face stacked dimer cation of these two species is presented as isomer A in Fig. 2. The H2CNH moiety is slipped from the center upon optimization (this complex has Cs symmetry), with the nitrogen pointing outwards at a right angle. At the MP2/aug-cc-pVTZ optimized geometry, the nearest carbon-carbon distance between the two moieties is 2.781, and the binding energy is 9.36 kcal mol-1 relative to the ethylene and methylene amine cation dissociation channel. Upon further single-point refinement at the CCSD(T)/aug-cc-pVTZ level of theory, the binding energy is 7.70 kcal mol-1. This relatively weak dimer complex is bound by the attraction of the diffuse π electron cloud and the positive charge of the closed shell methylene amine cation. This example shows how simple double-bonded species, including nitrogen containing ones, can form stacked complexes with small but non-negligible binding energies by electrostatic interactions when one monomer is charged. The [H2CCH2–H2CNH2]+ complex, Fig. 2.A, has several singlet-singlet electronic excitation bands in the UV; one around 4.62 eV (266 nm) has the strongest oscillator strength of 0.31. At the ωB97/cc-pVTZ level of theory the excitation energy for the same strong excitation band is 5.32 eV, which is much closer to the excitation energy, 5.62 eV, obtained at the EOM-CCSD/cc-pVTZ level of theory. The excitation energies calculated using B3LYP and ωB97 for all complexes studied are presented in Table 3. A more strongly bound four-membered cyclic structure, presented as B in Fig. 2, is also located, with 31.10 kcal mol-1 binding energy. This cyclic, slightly puckered structure is interesting as it incorporates a N atom in the ring, indicating the ease of ring formation in the gas phase due to electrostatic attraction. Which of these two product channels is obtained will depend to some extent on the relative orientation of the reactant partners as they reach association distance, though there will be a large thermodynamic driving force towards isomer B. Further association of ring structure B with acetylene might produce a six-membered carbon/nitrogen cyclic molecule.
moiety is slipped from the center upon optimization (this complex has Cs symmetry), with the nitrogen pointing outwards at a right angle. At the MP2/aug-cc-pVTZ optimized geometry, the nearest carbon-carbon distance between the two moieties is 2.781, and the binding energy is 9.36 kcal mol-1 relative to the ethylene and methylene amine cation dissociation channel. Upon further single-point refinement at the CCSD(T)/aug-cc-pVTZ level of theory, the binding energy is 7.70 kcal mol-1. This relatively weak dimer complex is bound by the attraction of the diffuse π electron cloud and the positive charge of the closed shell methylene amine cation. This example shows how simple double-bonded species, including nitrogen containing ones, can form stacked complexes with small but non-negligible binding energies by electrostatic interactions when one monomer is charged. The [H2CCH2–H2CNH2]+ complex, Fig. 2.A, has several singlet-singlet electronic excitation bands in the UV; one around 4.62 eV (266 nm) has the strongest oscillator strength of 0.31. At the ωB97/cc-pVTZ level of theory the excitation energy for the same strong excitation band is 5.32 eV, which is much closer to the excitation energy, 5.62 eV, obtained at the EOM-CCSD/cc-pVTZ level of theory. The excitation energies calculated using B3LYP and ωB97 for all complexes studied are presented in Table 3. A more strongly bound four-membered cyclic structure, presented as B in Fig. 2, is also located, with 31.10 kcal mol-1 binding energy. This cyclic, slightly puckered structure is interesting as it incorporates a N atom in the ring, indicating the ease of ring formation in the gas phase due to electrostatic attraction. Which of these two product channels is obtained will depend to some extent on the relative orientation of the reactant partners as they reach association distance, though there will be a large thermodynamic driving force towards isomer B. Further association of ring structure B with acetylene might produce a six-membered carbon/nitrogen cyclic molecule.
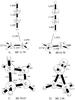 |
Fig. 1 MP2/aug-cc-pVTZ optimized geometry of the encounter complexes between HNCH+ and H2CCH2. Bond lengths are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented next to the figure labels. |
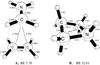 |
Fig. 2 Electrostatically bound closed shell π-complex and boat structure between the H2CCH2 and H2CNH |
III. [HCCH–HCNH]+ and [HCCH–HNCH]+.
Acetylene and HCNH+ are both observed in the ISM (Ziurys & Turner 1986). In total, three isomers that can be formed by barrierless association between HCCH and HCNH+ have been located on the singlet potential energy surface of the complex. Two T-shaped weakly bound complexes as well as a slightly more strongly bound cyclic isomer are presented in Fig. 3. In both T-shaped structures, shown as A and B in Fig. 3, the terminal H atoms of HCNH+ are attracted by the π electron cloud of HCCH. In HCNH+, the positive charge is distributed over all four atoms, but the N-terminal H atom has the most positive charge, hence the electrostatic attraction between that H atom and the π electron cloud of HCCH is stronger relative to the C-terminal H atom, and therefore the N-terminal H atom makes a stronger bond with the acetylene than the C terminal H-atom. The binding energies are presented in Table 1. The [HCCH–HCNH]+ complex is bound by 12.56 kcal mol-1 at the MP2/aug-cc-pVTZ level and 11.53 kcal mol-1 at the CCSD(T)/aug-cc-pVTZ level with respect to the HCCH and HCNH+ dissociation channel. Dissociating the more strongly bound [HCCH–HNCH]+ T-shaped complex into HCCH and HCNH+ will require 18.49 and 16.08 kcal mol-1 at the MP2/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ levels, respectively. As seen in Fig. 3, [HCCH–HNCH]+ has a shorter NH distance (1.895) relative to the CH distance, in [HCCH–HCNH]+, 2.110. Perhaps surprisingly, unlike the T-shaped complexes of HCNH+ and H2CCH2 discussed earlier (in Fig. 1), these complexes have weak electronic transitions in the ultraviolet. There is a cyclic isomer, presented in Fig. 3, with a four-membered ring and a slightly larger binding energy, relative to [HCCH–HNCH]+, of 20.75 kcal mol-1 at the CCSD(T)/aug-cc-pVTZ level. This is the simplest cyclic structure that incorporates a N atom into its framework. Calculated electronic excitation energies for all structures are presented in the Table 3. The difference in excitation energies between B3LYP (4.12 eV) and ωB97 (5.80 eV) DFT methods stems from the fact that B3LYP is not suitable for predicting excitations that have an associated charge transfer component. Of the two density functionals employed here, the ωB97 excitation energy comes much closer to that obtained by the EOM-CCSD/cc-pVTZ method (7.0 eV).
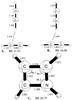 |
Fig. 3 Electrostatically bound closed shell π-complex and boat structure between the H2CCH2 and H2CNH |
IV. [HCN–H2CCH]+.
Another potentially viable mechanism for carbonaceous chain growth involves association of very abundant nitriles, such as HCN, and unsaturated molecular cations or fragment cations like vinyl cation. Steered by electrostatic ion-dipole attraction, vinyl cation can be associated with polar hydrogen cyanide, incorporating nitrogen in the chain growth process. Association of HCN with vinyl cation results in the formation of a strong covalently bound closed shell molecular cation. The MP2/aug-cc-pVTZ optimized geometry of this species, vinyl isocyanide cation, is shown in Fig. 4. Loss of a hydrogen from this complex leads to C3H3N+ which was found to form in the selected ion flow tube during UV induced dissociation of pyrimidine and may be a key species in pyrimidine formation (Bera et al. 2009; Fondren et al. 2007).
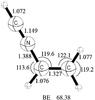 |
Fig. 4 Electrostatically bound closed shell π-complex and boat structure between the H2CCH+ and HCN, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
In vinyl isocyanide cation (see Fig. 4), the lone-pair electron on nitrogen donates into the vacant carbon π orbital to form a strong CN sigma bond. The central sp2 hybridized carbon makes a short single bond, 1.388, with the sp hybridized nitrogen. At the MP2/aug-cc-pVTZ level of theory the binding energy is 71.57 kcal mol-1 relative to the H2CCH+ and HCN dissociation channel. At the CCSD(T)/aug-cc-pVTZ level, the binding energy is 68.38 kcal mol-1 using the geometries optimized at the MP2/aug-cc-pVTZ level. The singlet ground state to singlet excited state vertical excitation energies obtained with TDDFT at the B3LYP/cc-pVTZ level of theory shows that the molecule has two relatively strong peaks in the UV at 8.35 eV (149 nm) and 5.76 eV (215 nm), with oscillator strengths of 0.11 and 0.30, respectively. The excitation energies (in eV) and oscillator strengths calculated at the B3LYP/cc-pVTZ and ωB97/cc-pVTZ levels of theories are included in Table 3, and are in quite good agreement with each other.
V. [HCN–HCC]+.
The neutral molecule acetylene (HCCH) and the acetyl radical (HCC) are observed species in the ISM, as is neutral C2. Protonated carbon CH+ has also been observed, however, HCC+ is as yet unobserved. Protonation of C2 will most likely produce HCC+ in the ISM, though the abundance of HCC+ in the ISM may be low due to its fast exothermic reaction with H2 (McWan et al. 1999). Reaction between two closed shell species, acetyl cation (HCC+) and hydrogen cyanide leads to two structural isomers: branched HCNC(H)C+ and linear HCCNCH+, whose MP2/aug-cc-pVTZ structures are presented in Fig. 5. A π complex between closed shell HCC+ and HCN is not stable due to very strong electrostatic attraction. The N lone pair of electrons in HCN can donate into the empty π orbital of the acetyl cation to make a strong sigma bond. The binding energies for these two isomers are very strong, 134.42 and 198.26 kcal mol-1, respectively at the same level of theory. Binding energies refined at the CCSD(T)/aug-cc-pVTZ level for the HCC+ and HCN dissociation channel are 154.30 and 205.59 kcal mol-1 for branched (Fig. 5) and linear isomers, respectively. The branched isomer has ten electronic absorption lines in the ultraviolet ranging from approximately 8.59 eV to 3.06 eV (144 nm to 405 nm). One of these bands at 6.39 eV (195 nm) exhibits a relatively strong oscillator strength of 0.36, while all others are weak. The linear isomer has only one very strong electronic excitation line at 166 nm with oscillator strength 1.1 computed at the TDDFT B3LYP/cc-pVTZ level of theory. Other excitations have nearly zero oscillator strength. For this case, the excitation energies and oscillator strengths computed with the B3LYP and range separated ωB97 functionals are in good agreement with each other (Table 3).
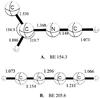 |
Fig. 5 Closed shell complexes of HCC+ and HCN or vice versa, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
VI. [HCCC–HCN]+.
The encounter between the HCCC+ ion and HCN leads to two structural isomers, presented in Fig. 6. A π complex of HCCC+ with HCN does not exist. However, the σ-bonded complexes are stable. The geometries optimized at the MP2/aug-cc-pVTZ level of theory are presented in Fig. 6. The binding energies, presented in Table 1, for the branched and the linear isomers are 40.42 and 55.03 kcal mol-1 respectively at the above mentioned level of theory. Binding energies relative to the HCCC+ and HCN dissociation channel further refined at the CCSD(T)/aug-cc-pVTZ level are 38.70 and 48.81 kcal mol-1 for the branched and the linear isomers (see Fig. 6). Both isomers have very strong electronic absorption signatures in the UV. One electronic excitation at 0.83 eV (B3LYP) or 0.89 eV (ωB97), as presented in Table 3, falls into the infrared region. Oscillator strengths and excitation energies for these complexes computed using TDDFT B3LYP/cc-pVTZ and ωB97/cc-pVTZ are presented in Table 3.
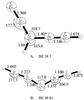 |
Fig. 6 Closed shell complexes of HCCC+ and HCN or vice versa, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure next to the labels. |
3.2. Open shell cations
VII. [H2CCH2–H2CNH]+.
The open shell cationic complex of H2CNH+ (H2CNH is an observed species in the ISM) (Gerakins 2011) with ethylene is an intriguing system. One proton linked to the carbon atom in H2CNH + is bound with the π-electron cloud of H2CCH2 in this cationic complex. The binding energy between H2CCH2 and H2CNH+ is 11.25 kcal mol-1 at the UMP2/aug-cc-pVTZ level (Table 2). The optimized geometry is presented in Fig. 7. ZAPT2/cc-pVTZ predicts the binding energy to be 9.44 kcal/mol and at the CCSD(T)/cc-pVTZ level, the binding energy is 10.28 kcal mol-1. The valence electronic excitation energies and oscillator strengths computed with the B3LYP/cc-pVTZ and ωB97/cc-pVTZ TDDFT methods are given in Table 3 and presented in Fig. 9 as a spectrum. The spectrum has multiple lines in the UV/VIS, only one of which at 2.27 eV (546 nm), has a relatively large oscillator strength of 0.23. Molecular orbital pictures indicate a charge transfer transition occurring from the highest doubly occupied molecular orbital (HDOMO) to the singly occupied molecular orbital (SOMO) (see Fig. 9) resulting in a very large oscillator strength at a very low wavelength for this electronic transition. For comparison, the excitation energies of the monomeric species are also presented in Table 4. It is clear that the low energy, intense electronic excitation band is a feature that is exclusive to the complex as there is no such band for either C2H4 or H2CNH+.
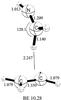 |
Fig. 7 Structure of weak electrostatically bound association complex of H2CNH+ with H2CCH2 molecule, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
VIII. [H2CNH2—H2CNH2]+.
Two isomers of the cationic dimer of H2CNH2 are presented in Fig. 8. One is a linear chain with carbon-carbon connectivity and the other is a π-stacked complex. The trans isomer, isomer A in Fig. 8, has an elongated 1.774 CC bond, and is bound by 38.42 kcal mol-1 at the UMP2/aug-cc-pVTZ level. Binding energies calculated using ZAPT2 and CCSD(T) with cc-pVTZ basis set are 39.22 and 40.20 kcal/mol respectively. The MP2/aug-cc-pVTZ optimized structure for isomer B (the π-stacked complex, see Fig. 8) shows that the carbon nitrogen distance between the monomer units is 2.622, which is an unusually long bond. The π-stacked complex is moderately strongly bound with a UMP2/aug-cc-pVTZ binding energy of 22.64 kcal mol-1. The binding energies predicted by the B3LYP (25.49) and ZAPT2 (23.71) levels of theory are also in agreement with that predicted by CCSD(T)/cc-pVTZ (24.98 kcal/mol). TDDFT calculations, Table 3, reveal electronic excitation energies with significant oscillator strength for both isomers due to charge-transfer type transitions. The trans isomer has a strong excitation transition at 3.01 eV (411 nm) with an oscillator strength of 0.19. The π-stacked isomer has a strong electronic excitation transition with 0.21 oscillator strength at 2.86 eV (433 nm).
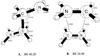 |
Fig. 8 The structures of the trans association complex and -stacked association complex of H2CNH2 with H2CNH |
3.3. Excitation energies
Excitation energies were calculated using time dependent density functional theory with two different functionals, the widely used B3LYP and ωB97 (Chai & Head-Gordon 2008), for comparison purposes. The electronic excitation calculations reported here refer to bands that would be observed in absorption from the ground electronic state to an excited state. Emission bands, which would likely be similar but shifted, can be calculated, but for this work we have limited the scope to absorption bands. Excitation energies and oscillator strengths of the complexes are presented in Table 3, and that of the constituting molecules and ions are presented in Table 4. For tightly bound covalent complexes, the excitation energies predicted by both the density functionals are in good agreement with each other. However, for the non-covalently bound complexes the electronic excitation energies predicted by B3LYP and ωB97 functionals can be different by more than 1 eV at times. As mentioned in the methods section, standard density functionals can perform very poorly for charge-transfer type excitations due to self-interaction errors, while range-separated hybrid density functionals, such as ωB97, have been shown to perform well in predicting excitation energies of charge transfer states. We have performed more accurate equations of motion coupled cluster singles and doubles (EOM-CCSD) calculations for the cases where the B3LYP and ωB97 excitation energies are found to be in disagreement. The EOM-CCSD calculations for the lowest five excited sates for the non-covalently bound complexes show very good agreement with those calculated using the ωB97 density functional.
We have computed the excitation energies of the five lowest energy excitations of the constituent neutral and ionic species and presented them in Table 4. A comparison of the excitation energies of the constituent monomer neutrals and ions with that of the complexes shows that none of the electronic excitations of the complexes are due to excitation in one constituent only. Some of the excitations are charge transfer in nature between the two moieties in the complex (e.g., Figs. 1.A,B) while others are due to complete reorganization of electronic structure after complexation. For example, first excitation energies of H2CCH2 and H2CNH, given in Table 4, are 7.89 eV and 8.04 eV respectively. The lowest excitation energy of the complex appears at much lower energy, 5.32 eV as given in Table 3, in the near ultra violet due to a charge transfer type transition between H2CCH2 and H2CNH. The electronic excitation energies produced by using the hybrid functional B3LYP and the range separated ωB97 agree well for all the species in Table 4 (unlike in the complexes where charge transfer plays a role). These excitation energies of the constituent monomer units can themselves be valuable for observation of these species in the ISM.
3.4. Astrophysical implications
The association complexes of mostly observed molecules and ions upon encounter are very interesting from an astrophysical perspective. As found in the present study (Tables 1 and 2), the ion-molecule complexes form with significant association energies that must be dissipated. In the low-density of the ISM, collisions are rare, and therefore, this excess energy is not readily dissipated to third bodies. The excess energy is then either lost radiatively or retained in the internal degrees of freedom of the complexes making the complexes vibrationally and rotationally hot. The weakly bound complexes may therefore have very short lifetimes and dissociate or isomerize to give rise to species other than their parents. However, in the current study we have not investigated dissociation rates for different channels. This could be investigated in the future. The open-shell cations have large oscillator strengths in the visible or near ultraviolet region. Therefore, the open shell nitrogen containing complexes, such as the one presented in Fig. 7 with strong electronic absorption lines in the visible (Fig. 9), are candidates for future laboratory study, and then observational detection. Most of the closed-shell association complexes have large oscillator strengths in the ultraviolet region of the electromagnetic spectrum. Linear conjugated carbonaceous cumulenes are common species in the ISM. Therefore, the linear and branched closed shell association ion-complexes, such as the ones presented in Figs. 4 − 6, should also be good candidates for future observation. From a growth perspective these nitrogen containing ionic association complexes can also serve as possible points for nucleation into larger molecules, such as nitrogeneous PAHs. The method of incorporation of nitrogen in the carbon framework is also related to how nitrogenous polycyclic molecules could form in the ISM. Identification of some of these intermediate complexes will help in understanding the involvement of nitrogen in the formation process of carbonaceous molecules in ISM.
 |
Fig. 9 The absorption spectrum (oscillator strength vs. wavelength (nm)) of the H2CCH2:H2CNH+ complex computed using time dependent density functional theory (TDDFT), at the B3LYP/cc-pVTZ level. The inset shows the highest doubly occupied molecular orbital (HDOMO) and the singly occupied molecular orbital (SOMO) of the complex that has a large oscillator strength. |
4. Conclusions
Gas phase ion-dipole encounter complexes of observed, and some yet unobserved, interstellar molecules have been studied using density functional theory, Møller-Plesset perturbation theory, and coupled cluster theory methods along with correlation consistent basis sets. Our main conclusion is that the simplest interstellar cations, steered by electrostatics, can initiate grain formation by forming larger chains, small cyclic cations, π-complexes, and strong ion-dipole complexes. In more detail, cations of some alkenes and alkynes, and nitrogen substituted alkenes and alkynes have been paired with their neutral counterparts making both closed shell and open shell complexes. Several different orientations, e.g. parallel, parallel displaced and T-shaped, of these encounter complexes have been tested for their stability, binding energy and electronic spectra. Acetylene and ethylene, when paired with protonated hydrogen cyanide (i.e. HCNH+), all of which are abundant in the ISM, make strong T-shaped closed-shell encounter complexes in which the proton plays a central role in bonding. Binding energies, ranging from 11.06 to 16.05 kcal mol-1, in these complexes are too strong to be a conventional hydrogen bond and too weak to be a conventional σ-bond. These examples include the T-shaped complexes in Figs. 1 and 3. Ethylene and H2CNH form a strong parallel-rotated π complex. Ethylene and H2CNH also form a more stable four-membered cyclic structure, though its binding energy is significantly less than a typical covalent bond. These examples are shown in Fig. 2. Closed shell π complexes of HCN and species with unfulfilled valencies (HCC+, HCCC+, and vinyl cation) are unstable upon geometry optimization and lead to σ bonded complexes with significant binding energies. These examples include Figs. 4 − 6. Open shell dimer cations of ethylene and H2CNH, and the complex of ethylene with H2CNH+ make stable and strong π complexes, Figs. 7 and 8.
To summarize, three types of complexes are predicted to form from the simple association of ions and molecules included in this study. Strong covalently bound molecules, ion-dipole bound complexes, and novel π-complexes of open shell cations are predicted. Most of the cationic species considered here have large oscillator strength electronic transitions in both the near and vacuum ultraviolet, and sometimes even in the visible region of the electromagnetic spectrum. Many of the electronic transitions with large oscillator strengths are due to charge-transfer type excitations between the monomer units, and are thus likely to be easily observed. The open-shell π-complexes, interestingly, have very large oscillator strengths in the visible region of the electromagnetic spectrum. This makes them possible candidates for some of the interesting observed interstellar emission features that extend throughout the UV-visible and well into the infrared electromagnetic spectrum.
All of the complexes studied here are formed by ion-molecule reactions without an association barrier. This point is supported by test ion-molecular trajectory calculations acetylene acetylene and ethylene, which will be reported in due course (Bera et al., in prep.). Thus, the question of which isomeric structure of a given complex is initially formed depends significantly on the encounter geometry of the ion-molecule species, though if there is a large difference in binding energies, there can be thermodynamic driving force towards the more stable complex. This is a question that we have not addressed in this study, but it would be interesting and desirable to perform ab initio trajectory calculations for a range of different impact factors and relative orientations for the complexes studied here to explore the initial isomer distributions. Subsequently, there can be internal rearrangements from one isomeric form to another, which are controlled by the excess internal energy and potential energy surface barriers. Hence, it would be useful to characterize the barriers to internal rearrangements in future work.
Online material
Appendix A: Cartesian co-ordinates (Angstrom) of the molecules, ions and complexes – reported here at the MP2/aug-cc-pVTZ level of theory
 |
Fig. A.1 a) C2H4–HNCH+. b) C2H4–HNCH+. c) C2H4–HNCH+, Boat. d) C2H4–HNCH+, Bridge. |
 |
Fig. A.2 a) C2H4–H2CNH |
 |
Fig. A.3 a) HCCH–HCNH+. b) HCCH–HNCH+. c) HCCH–HCHN+, Boat. |
 |
Fig. A.4 a) HCN–C2H |
 |
Fig. A.5 a) HCCNCH+, Branched. b) HCCNCH+, Linear. |
 |
Fig. A.6 a) HCCC(H)NC+, Branched. b) HCCCNCH+, Linear. |
 |
Fig. A.7 a) H2CCH2–H2CNH+. |
 |
Fig. A.8 a) |
![Mathematical equation: \hbox{$\rm {[}H_2CNH_2{]}_2^+$}](/articles/aa/full_html/2011/11/aa17103-11/aa17103-11-eq34.png)
Acknowledgments
P.P.B. acknowledges a fellowship award from the National Aeronautics and Space Administration (NASA) postdoctoral program administered by ORAU for NASA. T.J.L. gratefully acknowledge support from the NASA grant 08-APRA08-0050. The authors would like to thank the anonymous referee for constructive suggestions.
References
- Barinovs, G., & van Hemert, M. C. 2006, ApJ, 636, 923 [NASA ADS] [CrossRef] [Google Scholar]
- Bates, D. M., Anderson, J. A., Oloyede, P., & Tschumper, G. S. 2008, Phys. Chem. Chem. Phys., 10, 2775 [CrossRef] [Google Scholar]
- Becke, A. D. 1988, Phys. Rev. A, 38, 3098 [Google Scholar]
- Becke, A. D. 1993, J. Chem. Phys., 98, 5648 [NASA ADS] [CrossRef] [Google Scholar]
- Bera, P. P., Lee, T. J., & Schaefer, H. F., III. 2009, J. Chem. Phys., 131, 074303 [NASA ADS] [CrossRef] [Google Scholar]
- Black, J. H., Dalgarno, A., & Oppenheimer, M. 1975, ApJ, 199, 633 [NASA ADS] [CrossRef] [Google Scholar]
- Cami, J., Bernard-Salas, J., Peeters, E., & Malek, S. E. 2010, Science, 329, 1180 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Chai, J.-D., & Head-Gordon, M. 2008, J. Chem. Phys., 128, 084106 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Chillier, X. D. F., Stone, B. M., Salama, F., & Allamandola, L. J. 1999, J. Chem. Phys., 111, 449 [NASA ADS] [CrossRef] [Google Scholar]
- Dreuw, A., & Head-Gordon, M. 2004, J. Am. Chem. Soc., 126, 4007 [Google Scholar]
- Dunning, T. H. 1989, J. Chem. Phys., 90, 1007 [NASA ADS] [CrossRef] [Google Scholar]
- Fondren, L., McLain, J., Jackson, D., Adams, N., & Babcock, L. 2007, Int. J. Mass Spectrom., 265, 60 [NASA ADS] [CrossRef] [Google Scholar]
- Gerakins, P. 2011, http://science.gsfc.nasa.gov/691/cosmicice/interstellar.html [Google Scholar]
- Gerlich, D., & Horning, S. 1992, Chem. Rev., 92, 1509 [NASA ADS] [CrossRef] [Google Scholar]
- Halasinski, T. M., Weisman, J. L., Ruiterkamp, R., et al. 2003, J. Phys. Chem. A, 107, 3660 [CrossRef] [Google Scholar]
- Herbst, E. 2001, Chem. Soc. Rev., 30, 168 [CrossRef] [MathSciNet] [Google Scholar]
- Herbst, E., Roueff, E., & Talbi, D. 2010, Mol. Phys., 108, 2171 [NASA ADS] [CrossRef] [Google Scholar]
- Hirata, S., Lee, T. J., & Head-Gordon, M. 1999, J. Chem. Phys., 111, 8904 [NASA ADS] [CrossRef] [Google Scholar]
- Hirata, S., Head-Gordon, M., Szczepanski, J., & Vala, M. 2003, J. Phys. Chem. A, 107, 4940 [CrossRef] [Google Scholar]
- Hopkins, B. W., & Tschumper, G. S. 2004, J. Phys. Chem. A, 108, 2941 [CrossRef] [Google Scholar]
- Hudgins, D. M., Bauschlicher, C. W., Jr., & Allamandola, L. J. 2005, ApJ, 632, 316 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Jayatilaka, D., & Lee, T. J. 1992, Chem. Phys. Lett., 199, 211 [NASA ADS] [CrossRef] [Google Scholar]
- Johnson, D. R., & Lovas, F. J. 1972, Chem. Phys. Lett., 15, 65 [NASA ADS] [CrossRef] [Google Scholar]
- Lee, T. J., & Jayatilaka, D. 1993, Chem. Phys. Lett., 201, 1 [NASA ADS] [CrossRef] [Google Scholar]
- Lee, T. J., & Scuseria, G. E. 1995, in Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy, ed. S. R. Langhoff (Dordrecht, Germany: Kleuwer Academic Publishers), 47 [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Lee, T. J., Rendell, A. P., Dyall, K. G., & Jayatilaka, D. 1994, J. Chem. Phys., 100, 7400 [NASA ADS] [CrossRef] [Google Scholar]
- McWan, M. J., Scott, G. B., Adams, N. G., et al. 1999, ApJ, 513, 287 [NASA ADS] [CrossRef] [Google Scholar]
- Purvis, G. D., & Bartlett, R. J. 1982, J. Chem. Phys., 76, 1910 [NASA ADS] [CrossRef] [Google Scholar]
- Raghavachari, K., Trucks, G. W., Pople, J. A., & Head-Gordon, M. 1989, Chem. Phys. Lett., 157, 479 [NASA ADS] [CrossRef] [Google Scholar]
- Rhee, Y. M., Lee, T. J., Gudipati, M. S., Allamandola, L. J., & Head-Gordon, M. 2007, Proc. Natl. Acad. Sci., 104, 5274 [NASA ADS] [CrossRef] [Google Scholar]
- Salama, F., Galazutdinov, G. A., Krełowski, J., Allamandola, L. J., & Musaev, F. A. 1999, ApJ, 526, 265 [NASA ADS] [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Shao, Y., Molnar, L. F., Jung, Y., et al. 2006, Phys. Chem. Chem. Phys., 8, 3172 [CrossRef] [PubMed] [Google Scholar]
- Sinnokrot, M. O., & Sherrill, C. D. 2004, J. Phys. Chem. A, 108, 10200 [CrossRef] [Google Scholar]
- Small, D., Rosokha, S. V., Kochi, J. K., & Head-Gordon, M. 2005, J. Phys. Chem. A, 109, 11261 [CrossRef] [Google Scholar]
- Smith, D. 1992, Chem. Rev., 92, 1473 [CrossRef] [Google Scholar]
- Smith, I. W. M. 2002, Chem. Soc. Rev., 31, 137 [CrossRef] [Google Scholar]
- Snow, T. P. 2001, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 57, 615 [NASA ADS] [CrossRef] [Google Scholar]
- Snyder, L. E., & Buhl, D. 1971, ApJ, 163, L47 [NASA ADS] [CrossRef] [Google Scholar]
- Szczepanski, J., & Vala, M. 1993, Nature, 363, 699 [NASA ADS] [CrossRef] [Google Scholar]
- Weisman, J. L., Lee, T. J., & Head-Gordon, M. 2001, Spectrochim. Acta, 57, 931 [Google Scholar]
- Weisman, J. L., Mattioda, A., Lee, T. J., et al. 2005, Phys. Chem. Chem. Phys., 7, 109 [CrossRef] [Google Scholar]
- Werner, H.-J., & Knowels, P. J. 2006, MOLPRO, version 2006.1, a package of ab initio programs, H.-J. Werner, P. J. Knowles, F. R. Manby, M. Schütz, and others, see http://www.molpro.net [Google Scholar]
- Ziurys, L. M., & Turner, B. E. 1986, ApJ, 302, L31 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
Binding energies (in kcal mol-1) of the dimer cations between (mostly) known interstellar molecules and cations are listed.
Binding energies (in kcal mol-1) of the open shell complexes of astrochemically observed species.
TDDFT electronic excitation energies (in eV) of the complexes calculated with B3LYP/cc-pVTZ and ωB97/cc-pVTZ.
Excitation energies (in eV) and oscillator strengths of selected molecules and ions using time-dependent density functional theory (TDDFT) with B3LYP/cc-pVTZ and ωB97/cc-pVTZ.
All Figures
|
Fig. 1 MP2/aug-cc-pVTZ optimized geometry of the encounter complexes between HNCH+ and H2CCH2. Bond lengths are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented next to the figure labels. |
| In the text | |
|
Fig. 2 Electrostatically bound closed shell π-complex and boat structure between the H2CCH2 and H2CNH |
| In the text | |
|
Fig. 3 Electrostatically bound closed shell π-complex and boat structure between the H2CCH2 and H2CNH |
| In the text | |
|
Fig. 4 Electrostatically bound closed shell π-complex and boat structure between the H2CCH+ and HCN, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
| In the text | |
|
Fig. 5 Closed shell complexes of HCC+ and HCN or vice versa, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
| In the text | |
|
Fig. 6 Closed shell complexes of HCCC+ and HCN or vice versa, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure next to the labels. |
| In the text | |
|
Fig. 7 Structure of weak electrostatically bound association complex of H2CNH+ with H2CCH2 molecule, presented here at the MP2/aug-cc-PVTZ level of theory. Bond angles are expressed in Å and angles in degrees. Binding energies (BE, in kcal mol-1) are presented under the figure. |
| In the text | |
|
Fig. 8 The structures of the trans association complex and -stacked association complex of H2CNH2 with H2CNH |
| In the text | |
|
Fig. 9 The absorption spectrum (oscillator strength vs. wavelength (nm)) of the H2CCH2:H2CNH+ complex computed using time dependent density functional theory (TDDFT), at the B3LYP/cc-pVTZ level. The inset shows the highest doubly occupied molecular orbital (HDOMO) and the singly occupied molecular orbital (SOMO) of the complex that has a large oscillator strength. |
| In the text | |
|
Fig. A.1 a) C2H4–HNCH+. b) C2H4–HNCH+. c) C2H4–HNCH+, Boat. d) C2H4–HNCH+, Bridge. |
| In the text | |
|
Fig. A.2 a) C2H4–H2CNH |
| In the text | |
|
Fig. A.3 a) HCCH–HCNH+. b) HCCH–HNCH+. c) HCCH–HCHN+, Boat. |
| In the text | |
|
Fig. A.4 a) HCN–C2H |
| In the text | |
|
Fig. A.5 a) HCCNCH+, Branched. b) HCCNCH+, Linear. |
| In the text | |
|
Fig. A.6 a) HCCC(H)NC+, Branched. b) HCCCNCH+, Linear. |
| In the text | |
|
Fig. A.7 a) H2CCH2–H2CNH+. |
| In the text | |
|
Fig. A.8 a) |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.