| Issue |
A&A
Volume 520, September-October 2010
|
|
|---|---|---|
| Article Number | A64 | |
| Number of page(s) | 6 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/201014787 | |
| Published online | 04 October 2010 | |
Hyperfine structure in the rotational spectra of trans-formic acid: Lamb-dip measurements and quantum-chemical calculations![[*]](/icons/foot_motif.png)
G. Cazzoli1 - C. Puzzarini1 - S. Stopkowicz2 - J. Gauss2
1 - Dipartimento di Chimica ``G. Ciamician'', Università di Bologna, via Selmi 2, 40126 Bologna, Italy
2 -
Institut für Physikalische Chemie, Universität Mainz, 55099 Mainz, Germany
Received 14 April 2010 / Accepted 3 June 2010
Abstract
Context. Formic acid, HCOOH, is the simplest organic acid
and the first that has been identified in the interstellar medium.
Its astrophysical relevance has motivated this spectroscopic
study.
Aims. The aim of this investigation is to provide very accurate
rest frequencies for the trans isomer of HCOOH as well as to improve
the spectroscopic and hyperfine parameters available in the literature
for this molecule.
Methods. The Lamb-dip technique has been exploited in order to
record the rotational spectrum of trans-HCOOH at sub-Doppler resolution
in the millimeter- and submillimeter-wave frequency ranges and, when
possible, to resolve the hyperfine structure due to the hydrogen
nuclei. THz measurements have been carried out as well. The
experimental investigation has been supported by high-level
quantum-chemical calculations.
Results. As a consequence, we report here the most accurate
ground-state rotational parameters known so far for the main isotopic
species of trans-HCOOH as well as an accurate and reliable set of
hyperfine parameters.
Conclusions. The improvement in the accuracy of the ground-state
rotational and hyperfine parameters as well as of the rest frequencies
of rotational transitions might therefore be useful for future
radioastronomical observations.
Key words: methods: laboratory - techniques: spectroscopic - ISM: molecules - submillimeter: ISM - radio lines: ISM - molecular data
1 Introduction
Formic acid (HCOOH) exists in two rotameric forms, defined by whether
the two hydrogen atoms are cis or trans to each other. Both rotamers
are near prolate asymmetric tops with a planar structure
and ![]() symmetry (Winnewisser et al. 2002). The cis-rotamer lies approximately 1365(30) cm-1 (Hocking 1976)
higher in energy than the trans form; therefore, the
latter is about 800 times more abundant at room temperature. The
trans-HCOOH isomer is the subject of the present study, and we refer to
this form along the text, if not otherwise specified. To the
best of our knowledge, all radioastronomical observations reported in
the literature refer to the trans-rotamer of the main isotopologue.
symmetry (Winnewisser et al. 2002). The cis-rotamer lies approximately 1365(30) cm-1 (Hocking 1976)
higher in energy than the trans form; therefore, the
latter is about 800 times more abundant at room temperature. The
trans-HCOOH isomer is the subject of the present study, and we refer to
this form along the text, if not otherwise specified. To the
best of our knowledge, all radioastronomical observations reported in
the literature refer to the trans-rotamer of the main isotopologue.
The astrophysical relevance of formic acid stems from the fact that it is the simplest organic acid and the first observed in the interstellar medium (Zuckerman et al. 1971). It has been observed in various star-forming regions, in dense molecular clouds, etc.; we refer interested readers to Lattanzi et al. (2008) and references therein for an exhaustive list. We only point out that, as it is a species of intermediate complexity that shares common structural elements with biologically important species such as glycine, it represents a transition step between simpler species that are formed in the gas phase and complex hydrogenated species that are formed on grain surfaces (Turner et al. 1999).
As is well-known, astrophysical investigations require knowledge of accurate rest frequencies and/or accurate predictions for them. These are provided by laboratory measurements, and in this respect, sub-Doppler techniques such as the Lamb-dip method, whenever applicable, are the most suitable. In particular, the launch of the Herschel space observatory and the near-future operating international interferometer ALMA require knowledge of the transition frequencies of the molecules of astrophysical relevance in submillimeter-wave and THz regions with accuracies preferably better than 100 kHz (see, for example, Lattanzi et al. 2007, 2008). For these reasons, we report the investigation of the rotational spectrum of HCOOH in the 80-600 GHz frequency range recorded by means of the Lamb-dip technique, thus providing transition frequencies with an accuracy of 1 kHz, hence accurate spectroscopic parameters, as well as THz measurements performed with an accuracy of about 50-70 kHz. The Lamb-dip technique also allowed us to resolve the hyperfine structure of the spectrum due to the spin-rotation and spin-spin interactions of the two hydrogens.
The microwave and millimeter-wave rotational spectrum of formic acid has been studied since the 50s, but only quite recently Winnewisser et al. (2002) have reported measurements in the 835-993 GHz frequency range with an accuracy of 50-100 kHz and in the 1-2.9 THz frequency region with an uncertainty of 5 MHz. We also refer the reader to Winnewisser et al. (2002) for an exhaustive description of previous laboratory measurements. There is only one study of the hyperfine structure of the rotational spectrum reporting radio-frequencies measured with a molecular-beam spectrometer with an accuracy of 0.15 kHz (Chardon et al. 1976). Their analysis led to all the diagonal terms of the spin-rotation tensor for the two hydrogens, but only the two Caa constants were actually determined to a good accuracy. As explained later in the text, the spin-rotation constants of Chardon et al. (1976) provide erroneous predictions for the hyperfine structure in the millimeter-wave frequency range. More precisely, according to the values by Chardon et al. (1976), symmetric splittings of tens of kHz were expected in the millimeter-wave region, while smaller asymmetric splittings have actually been observed. To clarify this point, state-of-the-art quantum-chemical calculations of the hyperfine parameters involved have been carried out. As is made clearer later in the text, these computations allowed proper analysis and assignment of the spectra recorded.
2 Hyperfine structure of the rotational spectrum
The Hamiltonian describing the hyperfine structure of the rotational
spectrum of HCOOH can be given in terms of different contributions:
| (1) |
where
The spin-rotation Hamiltonian,
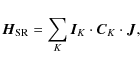
with K running over all nuclei with a non-zero nuclear spin
The dipolar spin-spin (Townes & Schawlow 1955; Flygare 1974) Hamiltonian,
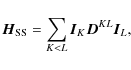
describes the dipolar interaction between two nuclear magnetic moments, namely
The various hyperfine components result from the
![]() ,
,
![]() = 0,
= 0, ![]() 1 selection rules, where F1 and F are the hyperfine quantum numbers originating from the coupling schemes
F1=J+IH1 and
F=F1+IH2.
1 selection rules, where F1 and F are the hyperfine quantum numbers originating from the coupling schemes
F1=J+IH1 and
F=F1+IH2.
3 Computational details
The experimental investigation was supported by quantum-chemical
calculations of the hyperfine parameters involved, namely, of the
spin-rotation and spin-spin interaction tensors. To ensure high
accuracy, all quantum-chemical calculations were performed using
state-of-the-art coupled-cluster (CC) techniques (Bartlett & Musia![]() 2007), together with large
atomic-orbital basis
sets. More precisely, equilibrium-value calculations were carried out
using the CC singles and doubles (CCSD) approach (Purvis & Bartlett
1982) augmented by a perturbative treatment of
triple excitations (CCSD(T); Raghavachari et al. 1989) in conjunction with the large cc-pCVnZ (n = Q,5) basis sets (Dunning 1989; Woon & Dunning 1995).
All electrons have been included in the electron-correlation treatment,
and the semi-experimental equilibrium geometry (Demaison et al. 2007)
was used as reference structure. Vibrational
corrections have also been considered as discussed in the following.
They were computed at the CCSD(T)/cc-pCVTZ level of theory. The CFOUR program package (2009) was used for all
calculations reported in the present work.
2007), together with large
atomic-orbital basis
sets. More precisely, equilibrium-value calculations were carried out
using the CC singles and doubles (CCSD) approach (Purvis & Bartlett
1982) augmented by a perturbative treatment of
triple excitations (CCSD(T); Raghavachari et al. 1989) in conjunction with the large cc-pCVnZ (n = Q,5) basis sets (Dunning 1989; Woon & Dunning 1995).
All electrons have been included in the electron-correlation treatment,
and the semi-experimental equilibrium geometry (Demaison et al. 2007)
was used as reference structure. Vibrational
corrections have also been considered as discussed in the following.
They were computed at the CCSD(T)/cc-pCVTZ level of theory. The CFOUR program package (2009) was used for all
calculations reported in the present work.
Let us start by considering the spin-rotation interaction, which requires computing the second-rank tensor
![]() .
Each element of the spin-rotation tensor has both an electronic and a
nuclear contribution. The electronic contribution can be evaluated as
the second derivative of the electronic energy with respect to the
rotational angular momentum
.
Each element of the spin-rotation tensor has both an electronic and a
nuclear contribution. The electronic contribution can be evaluated as
the second derivative of the electronic energy with respect to the
rotational angular momentum ![]() and the nuclear
spin
and the nuclear
spin
![]() (Flygare 1974). This is efficiently done using analytic second-derivative techniques (Stanton & Gauss 2000; Gauss & Stanton 1996,1997) in conjunction with
perturbation-dependent basis functions (Gauss et al. 1996);
the latter are often referred to as rotational London atomic orbitals
and were employed to improve basis-set convergence. On the
other hand, the nuclear contribution only depends on the geometrical
structure of the molecule.
(Flygare 1974). This is efficiently done using analytic second-derivative techniques (Stanton & Gauss 2000; Gauss & Stanton 1996,1997) in conjunction with
perturbation-dependent basis functions (Gauss et al. 1996);
the latter are often referred to as rotational London atomic orbitals
and were employed to improve basis-set convergence. On the
other hand, the nuclear contribution only depends on the geometrical
structure of the molecule.
The expression for the components of the dipolar spin-spin coupling tensor
![]() can be found, for instance, in Gordy & Cook (1984), and Abragam (1961). It is easily seen that the dipolar-coupling tensor
can be found, for instance, in Gordy & Cook (1984), and Abragam (1961). It is easily seen that the dipolar-coupling tensor
![]() is
completely determined once the molecular geometry is known, thus not
requiring any knowledge of the electronic structure of the molecule.
However, this strictly applies only to the case of a rigid,
non-vibrating molecule.
is
completely determined once the molecular geometry is known, thus not
requiring any knowledge of the electronic structure of the molecule.
However, this strictly applies only to the case of a rigid,
non-vibrating molecule.
Vibrational corrections to both spin-rotation and spin-spin tensors were evaluated with second-order vibrational perturbation theory (VPT2) (Mills 1972). The VPT2 approach has been described in detail in Auer et al. (2003) and already used to compute vibrational corrections for hyperfine parameters (see, for example, Cazzoli et al. 2009; Puzzarini et al. 2009; van der Tak et al. 2009). Within the VPT2 scheme, vibrational corrections are obtained by expanding the expectation value of the considered property over the vibrational wavefunction in a Taylor series around the equilibrium value with respect to normal-coordinate displacements. The required harmonic force field has been computed in a fully analytic manner (Gauss & Stanton 1997), while both the cubic force field and the corresponding derivatives of the considered property were obtained using finite-difference techniques as described in Stanton & Gauss (2000) and Stanton et al. (1998). The vibrational corrections, defined as the difference between the vibrationally averaged and equilibrium values determined at the same computational level, were then added to the best equilibrium results to derive theoretical best estimates for the ground-state data. The latter can be directly compared to the experimental values.
![\begin{figure}
\par\includegraphics[width=12cm,clip]{14787-fig1.eps}
\end{figure}](/articles/aa/full_html/2010/12/aa14787-10/img18.png)
|
Figure 1:
The hyperfine structure of the J = 182,16
|
| Open with DEXTER | |
4 Experimental details
For Lamb-dip measurements, a frequency-modulated, computer-controlled spectrometer in conjunction with a conventional free space cell, as shown in Cazzoli & Dore (1990a), was employed. To increase the sensitivity of the spectrometer as well as the Lamb-dip effect, the radiation path is doubled (Dore et al. 1999). A detailed description of the spectrometer is given in Cazzoli & Dore (1990b), whereas our experimental set up for performing sub-Doppler resolution spectroscopy has been described in some previous papers (see, for example, Cazzoli et al. 2002a,b, 2003). Therefore, here, we only report the main details relevant to the present investigation. The millimeter and submillimeter-wave sources employed are either frequency multipliers driven by Gunn diode oscillators or Gunn diodes themselves, covering, on the whole, the 80-600 GHz frequency range. The frequency modulation is obtained by sine-wave modulating the 73 MHz local oscillator of the synchronization loop at 1.666 kHz. A liquid He-cooled InSb detector was used, and its output was processed by means of a Lock-in amplifier tuned at twice the modulation frequency, so that the second derivative of the natural line profile was recorded. Those transitions that ended up too weak for observing the Lamb-dip effect were recorded at a Doppler-limited resolution.
In addition, Doppler-limited measurements were also carried out
in the 1.0-1.6 THz frequency range. In particular, the THz
frequency source is actually a tripler followed by another tripler
(RPG GmbH), and the overall radiation power is 10 to 40 ![]() W
(with also the twelfth harmonic detectable). As for the
millimeter-/submillimeter-wave spectrometer,
the frequency-modulation technique was employed, and the
80 MHz local oscillator of the synchronization loop is sine-wave
modulated at either 333 Hz or 10 Hz, depending on the
detector used. These are a liquid He-cooled Ge bolometer and a
Golay detector, and their output was processed by means of a Lock-in
amplifier tuned at twice the modulation frequency
(i.e., 666 Hz for the former, 20 Hz for the latter).
Another difference (with respect to the millimeter-submillimeter-wave
spectrometer) concerns the radiation
path. In fact, in the THz region, parabolic mirrors need to
be used for collimating and focalizing the radiation instead of high
density polyethylene lenses.
W
(with also the twelfth harmonic detectable). As for the
millimeter-/submillimeter-wave spectrometer,
the frequency-modulation technique was employed, and the
80 MHz local oscillator of the synchronization loop is sine-wave
modulated at either 333 Hz or 10 Hz, depending on the
detector used. These are a liquid He-cooled Ge bolometer and a
Golay detector, and their output was processed by means of a Lock-in
amplifier tuned at twice the modulation frequency
(i.e., 666 Hz for the former, 20 Hz for the latter).
Another difference (with respect to the millimeter-submillimeter-wave
spectrometer) concerns the radiation
path. In fact, in the THz region, parabolic mirrors need to
be used for collimating and focalizing the radiation instead of high
density polyethylene lenses.
For both Lamb-dip and THz measurements, the frequency sources are phase-locked to a rubidium frequency standard. The Lamb-dip measurements were always carried out at pressures lower than 0.5 mTorr. Such low values of working pressure have been chosen to minimize the dip widths as much as possible in view of improving the resolution, as well as to avoid pressure frequency shift. In addition, the source power was adequately reduced to have narrow and undistorted dips. The modulation depth used was adjusted in the range 5 to 15 kHz according to the experimental conditions and the transitions under consideration. Concerning Doppler-limited measurements, higher values of pressure (i.e., 20-30 mTorr and up to 50-90 mTorr in the THz region) and modulation depth (i.e., 300-400 kHz in the mm-/submm-wave region and 2.25-3 MHz in the THz frequency range) were employed. All measurements have been carried out at room temperature, using a commercial sample of trans-formic acid without any further purifications.
Going into more detail, about one hundred rotational transitions were recorded with an accuracy of about 1 kHz, and for most of them the hyperfine structure was resolved, while a total of about 150 rotational transitions were measured at Doppler-limited resolution in the submillimeter-wave and THz frequency regions. All transition frequencies are collected in Table S1 of the supplementary material.
![\begin{figure}
\par\includegraphics[width=11.5cm,clip]{14787-fig2.eps}
\vspace*{4mm}
\end{figure}](/articles/aa/full_html/2010/12/aa14787-10/img20.png)
|
Figure 2:
Line profile analysis of the hyperfine structure of the J = 221,21
|
| Open with DEXTER | |
5 Spectra analysis
The first step toward recording and analyzing the hyperfine structure
of the rotational spectrum of HCOOH was its prediction employing the
values for the spin-rotation constants available in
the literature. An example is provided by the bottom spectrum of
Fig. 1 for the J = 182,16
![]() 182,17 transition. The corresponding spectrum recorded by means of the
Lamb-dip technique is also depicted in Fig. 1,
which is the first from the top. At first sight, it is clear
that the two hyperfine structures differ significantly, with the former
characterized by a quartet with splittings as large as
182,17 transition. The corresponding spectrum recorded by means of the
Lamb-dip technique is also depicted in Fig. 1,
which is the first from the top. At first sight, it is clear
that the two hyperfine structures differ significantly, with the former
characterized by a quartet with splittings as large as ![]() 60-70 kHz
and the latter by a narrow triplet with two features not completely
resolved. As this was systematically observed for other
transitions as well, we resorted to quantum-chemical predictions for
the hyperfine parameters involved. Computed spin-rotation constants led
to the second spectrum from the bottom of Fig. 1.
While the extent of the hyperfine splittings is well reproduced, their
asymmetry is not. Only by accounting for the dipolar spin-spin coupling
(second spectrum of Fig. 1 from
the top), the recorded hyperfine structure is correctly predicted.
Therefore, after these first analyses, predictions of the hyperfine
structure for the other considered transitions were carried out based
on the computed spin-rotation and spin-spin interaction constants.
60-70 kHz
and the latter by a narrow triplet with two features not completely
resolved. As this was systematically observed for other
transitions as well, we resorted to quantum-chemical predictions for
the hyperfine parameters involved. Computed spin-rotation constants led
to the second spectrum from the bottom of Fig. 1.
While the extent of the hyperfine splittings is well reproduced, their
asymmetry is not. Only by accounting for the dipolar spin-spin coupling
(second spectrum of Fig. 1 from
the top), the recorded hyperfine structure is correctly predicted.
Therefore, after these first analyses, predictions of the hyperfine
structure for the other considered transitions were carried out based
on the computed spin-rotation and spin-spin interaction constants.
Once the spectra were recorded, the transition frequencies were
determined either by fitting the experimental data points to a
parabolic function or by a line profile analysis (see Cazzoli &
Dore 1990a), with the latter used
in the analysis of hyperfine structure involving partially blended
features and/or distorted dips. A graphical example of the line
profile analysis is provided
by Fig. 2. In all cases the
reported frequencies were obtained as averages of a set of
measurements, which have been found to be very reproducible.
In fact, in the case of Lamb-dip
measurements, the frequency differences for a given set are well within
1 kHz. Therefore, the latter can be considered a conservative
estimate of experimental uncertainties. For Doppler-limited
measurements, the frequencies were obtained only by fitting the
experimental data points to a parabolic function, and 20 kHz and
50 kHz (75 kHz in the case of detection with Golay) can be
considered conservative estimates for the experimental uncertainties in
the 500-600 GHz and THz frequency regions, respectively.
Figure 3 presents an example of THz measurements: a portion of the b Q band, with Ka = 9
![]() 8, is depicted. The good signal-to-noise ratio is also evident.
8, is depicted. The good signal-to-noise ratio is also evident.
![\begin{figure}
\par\includegraphics[width=15.5cm,clip]{14787-fig3.eps}
\end{figure}](/articles/aa/full_html/2010/12/aa14787-10/img22.png)
|
Figure 3:
Portion of the b Q band, with Ka = 9
|
| Open with DEXTER | |
Table 1: Rotational and hyperfine parameters of trans-HCOOH.
The analysis of the frequencies obtained in this work, as well as
those taken from the literature, were performed with an A-reduced
Watson Hamiltonian in the Ir representation (nearly prolate asymmetric top molecules (![]() = -0.95)).
= -0.95)).
6 Results and discussion
The retrieved frequency values are included in least-squares fits in which each line frequency has been weighted proportionally to the inverse square of its experimental uncertainty. Two different fits were carried out: the first one including our measurements together with the radio-frequencies (rf) recorded by means of a molecular-beam spectrometer by Chardon et al. (1976), and the second one also including the submillimeter-wave and THz measurements from Winnewisser et al. (2002). Both fits have been carried out with Pickett's SPCAT/SPFIT suite of programs (Pickett 1991). The results are reported in Table 1, where the present spectroscopic parameters are compared with those available in the literature as well as with our computed values.
Concerning the rotational parameters, the comparison of interest uses the data reported by Winnewisser et al. (2002). From Table 1, it is evident that, while the results of the two fits carried out in the present work agree within the standard uncertainties, discrepancies greater than the error bars are observed with respect to those from Winnewisser et al. (2002). Due to the accuracy of the Lamb-dip measurements carried out in the millimeter- and submillimeter-wave regions and thanks to the improved THz measurements (i.e., the uncertainties have been reduced from from 5 MHz to 50-75 kHz), we consider our rotational parameters (in particular, the rotational constants) more reliable.
It is evident from the results for hyperfine parameters reported in Table 1 why the results from Chardon et al. (1976) predict such overly large hyperfine structures, as seen in
Fig. 1. In fact, even if incorrectly determined, i.e., with uncertainties of about 50% or more, the Cbb and Ccc constants
are too large by about one order of magnitude, and they
also have the wrong sign in the case of the hydrogen linked to the
carbon atom. There are two comments that should be added. The first is
that, for each hydrogen, the sum of Cbb and Ccc constants given by Chardon et al. (1976)
is reasonably good, thus suggesting that the information they had at
hand is not sufficient for a proper determination of all the diagonal
terms. In this respect, it is noted that in the fit we had to
fix the ratio between the two Ccc constants
to the theoretical value, as they cannot be simultaneously
determined. The second remark is that the rf values by Chardon
et al. (1976) are reproduced very well by our fit (i.e., within the given uncertainties![]() ;
see Table S1 in supplementary material), hence by our hyperfine
parameters. Our comparison shows very good agreement between our
experimental results and the computed values, which explains why the
calculated parameters are able to predict the hyperfine structure of
the rotational spectrum, as shown in Fig. 1. Quantum-chemical calculations provide the full spin-rotation and spin-spin tensors, including the off-diagonal
elements. The latter have no relevance in predicting spectra as they cannot be determined experimentally.
;
see Table S1 in supplementary material), hence by our hyperfine
parameters. Our comparison shows very good agreement between our
experimental results and the computed values, which explains why the
calculated parameters are able to predict the hyperfine structure of
the rotational spectrum, as shown in Fig. 1. Quantum-chemical calculations provide the full spin-rotation and spin-spin tensors, including the off-diagonal
elements. The latter have no relevance in predicting spectra as they cannot be determined experimentally.
Finally, a comment on our calculated values is deserved. From our previous work (see, for example, Cazzoli et al. 2009; Puzzarini et al. 2009; Cazzoli et al. 2010), we already know that the CCSD(T) method employed in conjunction with a core-valence basis set of at least quadruple-zeta quality is suitable for obtaining equilibrium values for quantitative predictions, while a triple-zeta quality basis is usually sufficient to provide accurate vibrational corrections. Including vibrational corrections is mandatory for obtaining quantitative agreement with experiment. In fact, for both spin-rotation and spin-spin constants, vibrational corrections range from 0.06 kHz to 0.5 kHz, where the lower values apply to the smaller constants. The full set of computed data are collected in the supplementary material (see Table S2).
7 Conclusion
On the whole, we present the most accurate ground-state rotational parameters of HCOOH known so far. Since this molecule has a strong astrophysical relevance, we are convinced that the improved ground-state rotational parameters and rest frequencies obtained in the present investigation could be very useful for future observational purposes.
In the present investigation it is shown that, by employing the Lamb-dip technique, the hyperfine structure of rotational spectra can be recorded with high resolution, allowing determination of hyperfine parameters with good accuracy. Furthermore, the comparison between theory and experiment demonstrates that quantum-chemical computations can provide reliable reference values and, therefore if required, they can support, guide or even replace experimental determinations.
AcknowledgementsThis work has been supported in Bologna by ``PRIN 2007'' funds (project ``Trasferimenti di energia, carica e molecole in sistemi complessi'') and by the University of Bologna (RFO funds), and in Mainz by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
References
- Abragam, A. 1961, Principles of Nuclear Magnetism (New York: Oxford University Press) [Google Scholar]
- Auer, A. A., Gauss, J., & Stanton, J. F. 2003, J. Chem. Phys., 118, 10407 [NASA ADS] [CrossRef] [Google Scholar]
- Bartlett, R. J., & Musia▯, M. 2007, Rev. Mod. Phys., 79, 291 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., & Dore, L. 1990a, J. Mol. Spectrosc., 143, 231 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., & Dore, L. 1990b, J. Mol. Spectrosc., 141, 49 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., Dore, L., Cludi, L., Puzzarini, C., & Beninati, S. 2002a, J. Mol. Spectrosc., 215, 160 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., Dore, L., Puzzarini, C., & Beninati, S. 2002b, Phys. Chem. Chem. Phys., 4, 3575 [CrossRef] [Google Scholar]
- Cazzoli, G., Puzzarini, C., & Lapinov, A. V. 2003, ApJ, 592, L95 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., Puzzarini, C., Harding, M. E., & Gauss, J. 2009, Chem. Phys. Lett., 473, 21 [NASA ADS] [CrossRef] [Google Scholar]
- Cazzoli, G., Dore, L., Puzzarini, C., & Gauss, J. 2010, Mol. Phys., in press, [DOI: 10.1080/00268976.2010.484395] [Google Scholar]
- CF OUR A quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding, & P. G. Szalay, with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, O. Christiansen, M. Heckert, O. Heun, C. Huber, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. Matthews, T. Metzroth, D. P. O'Neill, D. R. Price, E. Prochnow, K. Ruud, F. Schiffmann, S. Stopkowicz, M. E. Varner, J. Vázquez, F. Wang, & J. D. Watts, and the integral packages MOLECULE, J. Almlöf, P. R. Taylor, PROPS, P. R. Taylor, ABACUS, T. Helgaker, H. J. Aa. Jensen, P. Jørgensen, & J. Olsen, and ECP routines by A. V. Mitin, & C. van Wüllen, For the current version, see http://www.cfour.de [Google Scholar]
- Chardon, J.-C., Genty, C., Guichon, D., & Theobald, J.-G. 1976, J. Chem. Phys., 64, 1434 [NASA ADS] [CrossRef] [Google Scholar]
- Demaison, J., Herman, M., & Liévin, J. 2007, J. Chem. Phys., 126, 164305 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Dore, L., Degli Esposti, C., Mazzavillani, A., & Cazzoli, G. 1999, Chem. Phys. Lett., 300, 489 [NASA ADS] [CrossRef] [Google Scholar]
- Dunning, T. H., Jr. 1989, J. Chem. Phys., 90, 1007 [NASA ADS] [CrossRef] [Google Scholar]
- Flygare, W. H. 1974, Chem. Rev., 74, 653 [CrossRef] [Google Scholar]
- Gauss, J., & Stanton, J. F. 1996, J. Chem. Phys., 104, 2574 [NASA ADS] [CrossRef] [Google Scholar]
- Gauss, J., & Stanton, J. F. 1997, Chem. Phys. Lett., 276, 70 [NASA ADS] [CrossRef] [Google Scholar]
- Gauss, J., Ruud, K., & Helgaker, T. 1996, J. Chem. Phys., 105, 2804 [NASA ADS] [CrossRef] [Google Scholar]
- Gordy, W., & Cook, R. L. 1984, Microwave Molecular Spectra, ed. A. Weissberger (New York: Wiley), 3rd edition [Google Scholar]
- Hocking, W. M. 1976, Z. Naturforsch., 31A, 1113 [Google Scholar]
- Lattanzi, V., Walters, A., Drouin, B. J., & Pearson, J. C. 2007, ApJ, 662, 771 [NASA ADS] [CrossRef] [Google Scholar]
- Lattanzi, V., Walters, A., Drouin, B. J., & Pearson, J. C. 2008, ApJS, 176, 536 [NASA ADS] [CrossRef] [Google Scholar]
- Mills, I. A. 1972, in Modern Spectroscopy: Modern Research, ed. K. N. Rao, & C. W. Matthews (New York: Academic) [Google Scholar]
- Pickett, H. M. 1991, J. Mol. Spectrosc., 148, 371 [Google Scholar]
- Purvis, G. D., III, & Bartlett, R. J. 1982, J. Chem. Phys., 76, 1910 [NASA ADS] [CrossRef] [Google Scholar]
- Puzzarini, C., Cazzoli, G., Harding, M. E., Vázquez, J., & Gauss, J. 2009, J. Chem. Phys., 131, 234304 [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Raghavachari, K., Trucks, G. W., Pople, J. A., & Head-Gordon, M. 1989, Chem. Phys. Lett., 157, 479 [NASA ADS] [CrossRef] [Google Scholar]
- Stanton, J. F., & Gauss, J. 2000, Int. Rev. Phys. Chem., 19, 61 [CrossRef] [Google Scholar]
- Stanton, J. F., Lopreore, C. L., & Gauss, J. 1998, J. Chem. Phys., 108, 7190 [NASA ADS] [CrossRef] [Google Scholar]
- Townes, C. H., & Schawlow, A. L. 1955, Microwave Spectroscopy (New York: McGraw-Hill) [Google Scholar]
- Turner, B. E., Terzieva, R., & Herbst, E. 1999, ApJ, 518, 699 [NASA ADS] [CrossRef] [Google Scholar]
- van der Tak, F. F. S., Müller, H. S. P., Harding, M. E., & Gauss, J. 2009, A&A, 507, 347 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Winnewisser, M., Winnewisser, P. B., Stein, M., et al. 2002, J. Mol. Spectrosc., 216, 259 [NASA ADS] [CrossRef] [Google Scholar]
- Woon, D. E., & Dunning, T. H., Jr. 1995, J. Chem. Phys., 103, 4572 [NASA ADS] [CrossRef] [Google Scholar]
- Zuckerman, B., Ball, J. A., & Gottlieb, C. A. 1971, ApJ, 163, L41 [NASA ADS] [CrossRef] [Google Scholar]
Footnotes
- ... calculations
- Tables S1 and S2 are only available in electronic form at the CDS via anonymous ftp to cdsarc.u-strasbg.fr (130.79.128.5) or via http://cdsarc.u-strasbg.fr/viz-bin/qcat?J/A+A/520/A64
- ... uncertainties
- Only the hyperfine components of the J =
94,5
 94,6 rotational transition have been
discarded as the corresponding
residuals are about 25 times greater than the given
uncertainties. A possible explanation is a typographical error
in Chardon et al. (1976).
94,6 rotational transition have been
discarded as the corresponding
residuals are about 25 times greater than the given
uncertainties. A possible explanation is a typographical error
in Chardon et al. (1976).
All Tables
Table 1: Rotational and hyperfine parameters of trans-HCOOH.
All Figures
|
|
Figure 1:
The hyperfine structure of the J = 182,16
|
| Open with DEXTER | |
| In the text | |
|
|
Figure 2:
Line profile analysis of the hyperfine structure of the J = 221,21
|
| Open with DEXTER | |
| In the text | |
|
|
Figure 3:
Portion of the b Q band, with Ka = 9
|
| Open with DEXTER | |
| In the text | |
Copyright ESO 2010
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.