| Issue |
A&A
Volume 550, February 2013
|
|
|---|---|---|
| Article Number | A127 | |
| Number of page(s) | 23 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/201220084 | |
| Published online | 06 February 2013 | |
Online material
Appendix A: Transmission probability computations
A.1. Eckart model
We compute the transmission probabilities of all the reactions involved in the water-producing and the methanol-producing networks by using the Eckart model (Eckart 1930; Johnston & Heicklen 1962). In this approach, an approximate potential energy surface (PES) is fitted as a function of the zero point energies (ZPEs) of the stationary points. The parameters needed for computing the transmission probability are the zero-point-corrected barrier heights of the forward and reverse reactions Vf and Vr, the frequency of the imaginary mode of the transition state νS, and the reduced mass of the reactants μ.
The Eckart potential can be parametrised as 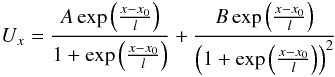 (A.1)where
(A.1)where 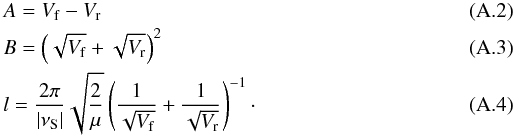 Once one has fitted the potential then the transmission probability, Pr, may be calculated using
Once one has fitted the potential then the transmission probability, Pr, may be calculated using  (A.5)where
(A.5)where 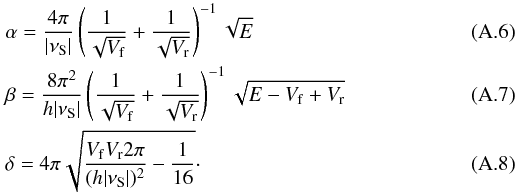 When the reactants are considered as excited by their formation involving an exothermic reaction, E refers to the excess energy of this reaction of formation. Otherwise, E is the thermal energy of the particles.
When the reactants are considered as excited by their formation involving an exothermic reaction, E refers to the excess energy of this reaction of formation. Otherwise, E is the thermal energy of the particles.
The Eckart model provides a significant improvement over square barriers but it can underpredict (or overpredict) the transmission probabilities of some reactions at low temperatures compared to more exact methods (see Peters et al. 2011). However, given the number of surface reactions considered in this work, exact quantum chemical computations for all reactions are not feasible.
A.2. CO2 formation
Based on the experimental work by Oba et al. (2010) who studied the formation of CO2 from CO and OH, CO2 is thought to be formed via the pathway 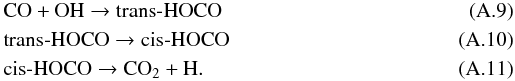 The electronic energy of the intermediate radicals (t-HOCO, c-HOCO) and the products (CO2 + H) are lower than for the reactants (CO + OH). Reactions involving t-HOCO and c-HOCO possess high activation barriers and/or are endothermic (Yu et al. 2001). Therefore, t-HOCO radicals continue to react only if their excess energy released by the chemical energy of reaction (A.9) is sufficient to overcome the high activation barriers of reactions (A.10) and (A.11). As in Goumans et al. (2008), HOCO radicals can also react with H atoms via barrierless reactions to form three pairs of products: H2 + CO2, H2O + CO, or HCOOH. The lack of more quantitative data leads us to assume a branching ratio of one to three for every product pair.
The electronic energy of the intermediate radicals (t-HOCO, c-HOCO) and the products (CO2 + H) are lower than for the reactants (CO + OH). Reactions involving t-HOCO and c-HOCO possess high activation barriers and/or are endothermic (Yu et al. 2001). Therefore, t-HOCO radicals continue to react only if their excess energy released by the chemical energy of reaction (A.9) is sufficient to overcome the high activation barriers of reactions (A.10) and (A.11). As in Goumans et al. (2008), HOCO radicals can also react with H atoms via barrierless reactions to form three pairs of products: H2 + CO2, H2O + CO, or HCOOH. The lack of more quantitative data leads us to assume a branching ratio of one to three for every product pair.
In fact, the energy released by reaction (A.9) absorbed by HOCO radicals can be transferred to the surface before reacting. However, the transfer rate of the chemical energy to the surface is very uncertain. The absence of formic acid HCOOH and the low abundance of HOCO radicals observed in the experiments of Oba et al. (2010) suggest that CO2 is readily formed from excited HOCO molecules. Therefore, most HOCO radicals continue to react before relaxing to their stable state. To reproduce these experiments, we assume that 99% of HOCO radicals are sufficiently excited to form CO2, whilst 1% of them are stabilized.
Goumans & Andersson (2010) performed gas-phase O-CO potential energy surface calculations, showing that O and CO can form a van der Waals complex, allowing O atoms to stay bound to CO for a long time. Following these results, we consider that O atoms that meet CO molecules (via direct accretion from gas-phase or surface diffusion) form a loosely bound O...CO complex. The H atoms that meet these O..CO complexes react via a barrierless reaction to form an excited HO...CO* complex. If the time for energy transfer to the surface is long enough, the complex can yield OH + CO, or t-HOCO* radical via barrierless reactions. Otherwise, the complex forms the t-HOCO radical through quantum tunneling. We consider that 99% of HO...CO* complexes continue to react without activation barriers.
Appendix B lists all the reactions involved in the formation of CO2 with their corresponding activation barriers and transmission probabilities.
A.3. Quantum chemical calculations
The OH + H2 reaction system has been theoretically studied by Nguyen et al. (2011). These authors have computed the forward and reverse reactions, the imaginary frequency of the transition states, and the rate constants of the eight reactions involving H2, HD, D2, OH, and OD using the semiclassical transition-state theory (SCTST).
The H2O2 + H reaction has been studied by Koussa et al. (2006) and Ellingson et al. (2007). However, data for the reactions involving deuterated isotopologues was not available. Therefore, to obtain the data required for the model, quantum chemistry calculations have been conducted with the Gaussian 09 (Frisch et al. 2009) program. For these calculations, the PBE0 (Perdew et al. 1996b,a; Adamo & Barone 1999) functional and the aug-cc-pVTZ (Dunning Jr 1989; Kendall et al. 1992) basis set were used because this combination produced results that are in good agreement with the experimentally determined values for the process in gas-phase (Klemm et al. 1975, who measured an activation barrier of 4.6 kcal/mol = 2300 K) .
The CO + H reaction has been theoretically studied by several authors (Woon 2002; Andersson et al. 2011; Peters et al. 2012). We decided to use the work of Peters et al. (2012) because they used the most accurate methodology and obtained a value for the activation energy for the formation of HCO that best agrees with the gas-phase experiment (Wang et al. 1973). The transmission probabilities of all the reactions producing deuterated formaldehyde and methanol are computed from this reaction and relative rates measured by Nagaoka et al. (2007) and Hidaka et al. (2009) or deduced by TCK12b.
The formation of carbon dioxide includes two reactions having an activation barrier. Quantum calculations of Talbi et al. (2006), Goumans et al. (2008), and Goumans & Andersson (2010) showed that reaction (11) has an activation barrier of 2500 − 3000 K, leading to a transmission probability
of 5 × 10-23 (Goumans & Andersson 2010; Garrod & Pauly 2011). Assuming an activation energy of 2500 K and considering a square barrier, we reproduce this transmission probability with a barrier width of 0.8 Å. The transmission probabilities of the reaction pathways involved in reaction (12), and including HOCO radicals, the van der Waals complex HO...CO, and their deuterated isotopologues, were deduced from the potential energy surface computed by Yu et al. (2001). These authors computed the stationary points of the potential energy surface of this reaction using an extrapolated full coupled cluster/complete basis set (FCC/CBS) method.
Appendix B: List of grain surface reactions
List of grain surface chemical reactions considered in this work along with their activation barriers and transmission probabilities.
© ESO, 2013
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.