| Issue |
A&A
Volume 689, September 2024
|
|
|---|---|---|
| Article Number | A118 | |
| Number of page(s) | 11 | |
| Section | Interstellar and circumstellar matter | |
| DOI | https://doi.org/10.1051/0004-6361/202449701 | |
| Published online | 06 September 2024 | |
Thermodynamical stability of [CNN and NCN] sequences as indication of most abundant structures in the ISM
1
Univ Rouen Normandie, INSA Rouen Normandie, CNRS, Normandie Univ, COBRA UMR 6014, INC3M FR 3038, F76000 Rouen, France and CNRS, Sorbonne Université,
LCT UMR 7616,
75005
Paris,
France
2
Institut Parisien de Chimie Physique et Théorique IP2CT (FR2622), Sorbonne Université,
Paris,
France
3
Univ Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS,
ISCR UMR 6226,
35000
Rennes,
France
e-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
22
February
2024
Accepted:
13
June
2024
Abstract
Context. Most of the molecules identified in the interstellar medium (ISM) are organic compounds and more than 50 have one isomer or more. Statistically, the most stable isomer of a given chemical formula is the most abundant. This occurrence is verified up to ~90% of the detected species leading to the so-called minimum energy principle (MEP).
Aims. Our main objective is to increase the list of the 14 bis-nitrogen species already detected. We focus on ten CxHyNz isomer families with x = (1, 2, 3), y = (0, 2, 4, 6, 8), z = 2. To this end, we look for a reliable and economic way to provide energy scales.
Methods. We employed standard quantum chemistry methods to determine the relative position of each isomer on the energy scales of each family. We systematically applied density functional theory (DFT) treatments using basis sets of increasing size and quality (6-311++G** and cc-pVQZ). When reasonably feasible, we then performed high-level coupled cluster calculations (CCSD) using the same basis sets to refine relative energies.
Results. All 14 bis-nitrogen species already identified in the ISM indeed satisfy the MEP. We determine the relative thermodynamic stability of the isomers with a CxHyN2 formula of each of the ten sets (94 compounds altogether), and hightlight those that are potentially detectable. By increasing the number of carbon atoms, we find 15 compounds that are by far the most stable candidates.
Conclusions. We confirm that, within the limits of thermodynamics, MEP is an efficient and easily applicable tool for identifying the isomers in a given series that have a greater probability of being detected. Computationally, the combination “B3LYP/cc-pVQZ” provides a suitable compromise for determining energy differences and dipole moments. Clearly, the isomers containing the [NCN] sequence should be prioritized over those with [CNN] in future observation campaigns.
Key words: astrochemistry / ISM: abundances / ISM: molecules
© The Authors 2024
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
Over 300 molecules have been identified in the interstellar medium (ISM) and many are organic compounds1. Most of those detected contain between 2 and 13 atoms with a significant decrease in population for each additional atom starting from 3 atoms. There has been a steady increase in the number of compounds detected with more than 5 atoms (complex organic molecules (COMs)) and each new detection provides crucial information about the chemistry of the object in which it is identified. For any compound with a dipole moment, its detection is usually achieved via the comparison between the microwave or millimeter spectrum recorded in the laboratory and that obtained from observations of molecular clouds, some of which contain thousands of unassigned bands.
It should be stressed that the spectra of many candidate species have never been recorded, and for some of these, synthesis in the laboratory has not yet been carried out. Due to the very small quantity of compounds detected compared to the number of theoretically possible molecules, it is necessary to find techniques or rules of thumb allowing the number of candidates to be significantly reduced. The most common ways for selecting compounds to search for in the ISM are based on:
- (i)
analogy with those already detected,
- (ii)
targeting the plausible missing pieces in accepted chemical networks in the molecular clouds,
- (iii)
energetic data derived from phenomenological models, such as the minimum energy principle (MEP).
This latter principle hightlights that most compounds identified in the ISM correspond to the thermodynamically most stable isomers (Lattelais et al. 2009, 2010b). Thus, in order to help select new candidates for observation, we suggest the following guidelines:
- (i)
selection of a formula with no isomer detected in the ISM,
- (ii)
selection of derivatives by replacement of an atom by an isovalent atom down the periodic chart (O by S; N by P; C by Si, etc.) or by an isoelectronic group (O by NH, etc.) or any combination of the two,
- (iii)
selection of the lowest-energy isomers of the new series thus obtained (most likely to be present in ISM according to the MEP).
Beyond these energetic considerations, it is important to consider that the intensity of the microwave or millimeter spectrum of any molecule is proportional to the square of its electronic dipole moment (μ2). Today, there are 106 detected molecules according to the current knowledge containing at least one nitrogen atom (see Table A.1). Only 14 contain two nitrogen atoms (Table 1), and no molecule with three has yet been detected. Thus, compounds with two nitrogen atoms were selected as new targets for future observations. Here, we report a study of systems with formulae of CxHyN2 with x = (1, 2) combined with y = (0, 2, 4, 6, 8). The number of hydrogens added is determined so as to obtain full saturation of all families containing backbones with one or two carbons in a first step.
The list of compounds detected with two nitrogen atoms (Table 1) very recently increased with the identification of N-cyanomethanimine H2C=N-CN (San Andrés et al. 2024). The largest compounds detected containing two nitrogen atoms, aminoacetonitrile (Belloche et al. 2008) and urea (Belloche et al. 2019) are composed of eight atoms. These latter are important intermediates in prebiotic syntheses, and so the search for species of formula C3HyN2 (y = 2, 4) is an interesting and congruent path worthy of exploration. The main challenge in this type of survey is to design a credible and comprehensive approach for selecting a set of candidates for each of the series of molecules. The investigations reported here focus on neutral species only, avoiding free radicals with one unpaired electron or any positive ion2.
In summary, we consider the ten following series of isomers: CN2 (one singlet state and one triplet state for each of the 2 isomers), CH2N2 (6 isomers), CH4N2 (8 isomers), CH6N2 (2 isomers), C2N2 (3 isomers), C2H2N2 (6 isomers), C2H4N2 (15 isomers), C2H6N2 (13 isomers), C2H8N2 (6 isomers), C3H2N2 (8 isomers), C3H4N2 (23 isomers), which amounts to 94 molecules altogether.
For each CxHyN2 isomer, diastereomers (Z) and (E) are considered in this work. In such an exhaustive study, it is necessary to reach a compromise between level of theory and calculation requirements.
Molecules detected in space so far containing two nitrogen atoms as a function of the total number of atoms (the chronological order of detection is shown in parenthesis).
2 Computational logic
This series of calculations is first organized so as to form a simple protocol that can be applied to a large panel of systems. Thus, this method aims to identify molecular structures that could initiate laboratory work in view of future observations. To this end, we rely on recent studies showing close relations between highlevel numerical simulations, and detections or nondetections of molecular species. Among these, are works on heteroaromatic rings (Lattelais et al. 2010a), HxCyOz (Karton & Talbi 2014), urea/thiourea (Fourré et al. 2016), methylketene (Bermúdez et al. 2018), and [C,N,O] fragments (Fourré et al. 2020).
Before systematic application, we must check the consistency between the energy scales, regardless of the computational model. Indeed, the energetic ordering of the isomers is compared through the extension of the one-particle space (atomic basis set) and the n-particle space (configuration space) for each family of isomers. And if so, a reliable estimate of the possibility of detection of a compound of larger formula as C3H4N2 is conceivable following the B3LYP/cc-pVQZ level calculations. This approach will avoid large-scale coupled-cluster investigations that become rapidly intractable for such large systems. B3LYP was chosen as functional because it led to relevant results in previous studies for geometry determination (Becke 1993).
To establish the consistency between the energy scales, we developed a multi-step strategy to reliably position each isomer on these scales for the ten series explored in this study. For this strategy, we used two basis sets of improved quality, namely 6-311++G** and cc-pVQZ (hereafter referred to as (B1) and (B2) respectively), for all DFT calculations at the B3LYP level (Becke 1993; Lee et al. 1988). We verified that each structure is an energy minimum by carrying out a vibrational analysis (Stephens et al. 1994)3.
In order to position compounds on the energy scales, the relative stabilities of all isomers of smaller dimensions with one and two carbon atoms (up to C2H2N2) were recomputed at higher levels of theory using the same basis sets. More precisely, we fully reoptimized the geometries at the highly correlated CCSD level of theory, in which the electronic correlation is calculated by including the interaction between the ground state and the excited states obtained by single and double excitations. The zero point vibrational energies (ZPEs) are calculated at that same level of theory.
From C2H4N2 molecules and larger, that is, C2HyN2 (y=6,8) and the series C3H2N2, we carried out these calculations at the DFT(B1) and DFT(B2) levels, and only at the DFT(B2) level for C3H4N2 isomers. All energy differences reported include ZPE corrections4. We verified that including entropic terms to determine Gibbs free energies of formation at the temperature estimated for the considered ISM region has a marginal impact on the values of the energy differences.
It should be noted that dipole moments extracted from a CCSD(B2) calculation are actually performed using a computational procedure based on a quadratic configuration interaction that relies on all possible single and double excitations from the ground state. In the course of this study, we began to question whether or not the role of diffuse functions in basis sets is possible source of computational bias. On selected model candidates, we compared the results with and without diffuse atomic functions in the basis sets, that is (B0) with (B1) and (B2) with (B3). These results are presented in Table C.1. As in the series built around one, two even three carbons: C2N2, C2H2N2, C3H2N2, these comparisons show that incorporation of diffuse functions has very little influence on the energy scales and dipole moments. The following tables gather the calculated energies and dipole moments and are organized as energy scales starting from the most stable species of the considered family of CxHyN2 isomers (x = (1, 2, 3), y = (0, 2, 4, 6, 8)). All the calculations were carried out using methods and basis sets as implemented in the Gaussian 09 and Gaussian 16 software packages (Frisch et al. 2009, 2016).
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing CH(y=0,2,4,6)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
3 From N=C=N to H2N−CH2−NH2
The number of compounds with a unique carbon and two nitrogen atoms (Table 2) can be partitioned in four subclasses according to the number of additional hydrogen atoms completing the structure.
3.1 CN2 species
Two molecules of different geometries, cyanonitrene NCN, and diazomethylene CNN, were characterized in the laboratory by Wasserman, Barash & Yager (1965) using electron paramagnetic resonance spectroscopy, a technique that is not really applicable to observations in space. Both have a triplet ground state, with NCN being the most stable. The singlet-triplet separation is on the order of ~28–33 kcal mol−1 for NCN and CNN. On the whole, the energy scales present the same ordering for all the levels of calculations. No CN2 molecules are detected in the ISM but, to our knowledge, their microwave or millimeter spectra have never been recorded in the laboratory. Without a dipole moment and therefore without a millimeter spectrum, the detection of NCN isomers remains a real observational challenge (Table 2A).
It should be noted that the values of the energy differences and dipole moments obtained at DFT(B2) are close to those obtained for the single point CCSD/DFT(B2), which is also close to the full CCSD(B2), regardless of the spin state singlet, or triplet5.
3.2 CH2N2 species
Six CH2N2 molecules of different geometries are stable, all in a singlet state (see first lines in Table 2B). The lowest two in energy carbodiimide and cyanamide (HN=C=NH and NC-NH2), are almost equivalent at the DFT level, with HN=C=NH being about 1 kcal mol−1 lower than NC-NH2. Introducing electronic correlation at the CCSD level produces the opposite result, with NC–NH2 being more stable than HN=C=NH by ~4 kcal mol−1. Regardless of the method, the dipole moment values remain steady, and the final result is consistent between DFT and correlated CCSD calculations (4.5 versus 2.0 Debye).
The following, third isomer diazomethane CH2NN(diazo), which can be also represented by a zwitterion H2C=N+=N−, is well above first two on the energy scale.
The last three isomers CH2NN(cycle), H2N−NC and HCNNH(cycle) are much higher on the energy scale for two different reasons: the strong tensions in the three-membered (2H-and 1H-diazirine) rings for the fourth and sixth isomers, and the N-N weak bond between the amino and the -NC isocyanic groups for the isocyanamide.
Carbodiimide and cyanamide have already been observed in the ISM (McGuire et al. 2012 and Turner et al. 1975). The microwave spectra of the other isomers have been recorded (Pierce & Dobyns 1962; Schäfer et al. 1981), except for the sixth, 1H-diazirine, for which the rotational constants have only been calculated (Bera et al. 2022). Cyanamide was detected more than 30 years before carbodiimide. This could be explained by the relative abundances of the two molecules: carbodiimide exhibits a ten times lower abundance than cyanamide in the cloud where both have been detected (Ramal-Olmedo et al. 2023; Zhang et al. 2023).
In such cases, where energy differences are within a few kcal mol−1 and susceptible to reverse with the level of correlation used, it should be noted that dipole moments values are relatively steady. Therefore, it could be appropriate to analyze correlation effects (CCSD) to draw up the energy scale able to reproduce the right ordering of isomers. In some cases, the dipole moments might improve the ease of detection.
It is also interesting to note that the energetic results and dipole moments at the single point CCSD/DFT(B2) are still very close to those of full CCSD(B2).
3.3 CH4N2 species
Eight isomers follow, formally from the addition of four hydrogen atoms to the (cyanonitrene) NCN and (diazomethylene) CNN backbones (Table 2C). The two most stable isomers are (Z)- (cis) and (E)- (trans) formamidine, which are close in energy at both DFT and CCSD levels (~2 kcal mol−1). In between, we find the H3N−HCN complex, whose microwave spectrum was obtained by Fraser et al. (1984). This complex could be stable in the ISM at the temperature of 10 K. This ordering for the three first isomers is only found in correlated calculations, and not in DFT.
The synthesis of unsubstituted formamidine (HN=CH–NH2) has never been reported. Methyldiimides (also called methyldiazenes, H3C-N=NH), diastereomers (E) and (Z) are about 30 kcal mol−1 higher on the energy scale at the sixth and seventh positions. As shown in the Section 3.1, singlet and triplet states are stable structures. However, in that case, these electronic states are found only for the symmetrical geometry of the H2N-C-NH2 isomer at the fourth and eighth positions. For this diaminocarbene, the singlet is more stable than the triplet, contrary to the NCN nonhydrogenated molecule, with a singlet-triplet separation of approximately 50 kcal mol−1. None of the isomers with a CH4N2 formula have been detected in the ISM. For the (E)-methyldiimide, the microwave spectrum of the deuterated derivative (CH3N2D) has been reported, the N-D bond giving a stronger stability to this isotopolog in comparison with the hydrogenated isomer (Steinmetz 1970).
On the computational side, we note that the dipole moment does not change significantly between each particular energy scale, regardless of the level of calculation. In other words, the energetic ordering of the isomers remains the same, and does not depend on the computational model for all neutral compounds. Nevertheless, the second position of the H3N−HCN complex on this energy scale requires a more in-depth treatment of electronic correlation. Including the correlation effect at the CCSD level provides very little difference between the CCSD/DFT(B2) single point and the fully optimized CCSD(B2) system.
3.4 CH6N2 species
Only two isomers fulfill the saturation requirement in this series (Table 2D). These compounds, methanediamine (H2N−CH2−NH2) and methylhydrazine (H3C−NH−NH2), are very different regarding their chemical functions. Moreover, isomerization which implies a profound atomic rearrangement, is difficult, and so these species should originate from different precursors. Following the MEP, the energy difference favors methanediamine (H2N−CH2−NH2). Here again, the DFT(B2), CCSD/DFT(B2) and CCSD(B2) lead to similar results.
4 From NC-CN to H2N−CH2−CH2−NH2
4.1 C2N2 species
Neglecting high-energy cyclic isomers, only three compounds are worthy of consideration (Table 3A). Two of these, the cyanogen NCCN and diisocyanogen CNNC species have no electric dipole moment for symmetry reasons. As a consequence, the most stable isomer, NCCN, cannot be identified by its rotational spectrum. However, the presence of this molecule is asserted by the detection of the corresponding protonated molecule NCCNH+ (Agúndez et al. 2015). The only detected (Agúndez et al. 2018) NCNC (also written CNCN) asymmetric species is second on the energy scale, at around 24 kcal mol−1 higher than the lowest symmetric isomer. The third isomer, CNNC, at approximately 71 kcal mol−1 above the most stable isomer (NCCN), with no dipole moment has very little chance of being detected. For the C2N2 family of isomers, we can consider two detections; one direct, namely that of the asymmetric compound, and one indirect, that of the symmetric compound, NCCN, whose presence is only deduced from its protonated form in the ISM. However, a comparison between the abundances of the protonated adduct and that of the neutral species is not relevant.
4.2 C2H2N2 species
The six lowest-energy isomers of C2H2N2 formula are presented in Table 3B. In DFT, the two first isomers with the lowest energies are diastereomers Z (cis) and E (trans) of iminoacetonitrile (also called C-cyanomethanimine). These contain a cyano group whose triple bond is conjugated with an imine CH=NH structure. Here, (Z)-iminoacetonitrile is more stable than its (E)-diastereomer and is reported to be the most abundant by Rivilla et al. (2019), which is in good agreement with the MEP. In third position, the isomer N-cyanomethanimine H2C=N−CN shows some energy differences in the range of ~4 to 6 kcal mol−1 for the two most stable compounds, according to the level of correlation. In fourth and five positions (Table 3B) on the other hand, the cumulene ((Z)-HN=C=C=NH)) is less stable than its (E)-diastereomer. However, the energy differences are so small and none diastereomer should be eliminated for energetic reasons. By contrast, the (E)-diastereomer with no dipole moment can be discarded for detection by microwave spectroscopy. Thus, among the isomers with a C2H2N2 formula, the two first have been detected in the ISM (Zaleski et al. 2013; Zhang et al. 2020). Regarding the third, its microwave spectrum is already known (Evans et al. 1991; Winnewisser et al. 1984) and this N-cyanomethanimine was very recently identified in the Galactic center source G 0.693–0.03 with a low abundance (San Andrés et al. 2024).
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=0, 2)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
4.3 C2H4N2 species
We selected 15 isomers (Table 4C). The first is the simplest α-aminonitrile, a precursor of the simplest α-amino acid glycine through hydrolysis. Next are N-methylcarbodiimide and N-methylcyanamide, both of which exhibit thermodynamic stabilities not far from the most stable 2-aminoacetonitrile. Following these, are a series of diimines (ethanediimine and N-methylene-methanimidamide) whose thermodynamic stabilities slowly deviate from those of the three most stable C2H4N2 isomers. The diastereomers of two diimines are positioned from fourth to eighth isomer on the energy scale (Table 4C). Only the thermodynamically most stable compound, the 2-aminoacetonitrile, has been detected (Belloche et al. 2008). The microwave spectrum of the third isomer, N-cyanomethylamine, has been recorded (Bak & Svanholt 1980a), but we failed to find microwave spectra for other isomers. Therefore, it is quite difficult to conclude on the efficiency of the MEP in this series given the lack of spectroscopic data for these compounds.
4.4 C2H6N2 species
Among the 13 isomers considered in this series, ten are within the energetic range of ~30 kcal mol−1 over the most stable isomer, (Z)-N-methyl-methanimidamide, HN=CH-NH-CH3 as shown in Table 5D.
The ordering of isomers on the energy scale shows that (Z)-HN=CH-NH-CH3 lies a little less than 1 kcal mol−1 below the second isomer, ethene 1,1-diamine. In third position, the (E)-diastereomer of N-methyl-methanimidamide is less stable, with only a difference in energy of ~1 kcal mol−1. The presence of these three isomers within a range of 1.5 kcal mol−1 makes the identification based on relative energies quite problematic. In addition, this second isomer (H2N)2C=CH2, an isoelectronic of urea (H2N)2C=O, is not the most stable isomer of the series here. It demonstrates the danger of using the isoelectronic criterion for building new series supposed a priori to satisfy the MEP. Nevertheless, taking into account that these three most stable isomers have dipole moments of ~1 or 3 Debye, the probability of detection of N-methyl-methanimidamide, either (E) or (Z), is multiplied by close to an order of magnitude at equivalent abundance.
Higher on the energy scale (Table 5D), between 7.5 and 9.5 kcal mol−1 from the most stable (Z)-N-methylmethanimidamide, are the compounds (Z)- and (E)-ethene-(1,2)-diamine and 2-iminoethanamine, in which the two nitrogens are separated by two carbon atoms. From the 9th position on this energy scale to the 13th and last, all isomers contain the much less stable [CNN] sequence. In this way, (E)- and (Z)-dimethyldiazene at 27 and 36.5 kcal mol−1 from the lowest energy isomer, present a −N=N− linkage, generally a weak point of a molecular backbone. The first cycle, the N-amino-aziridine ranks in the last position, at almost 65 kcal mol−1 and well above the most stable, which makes detection quite improbable.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=4)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=6,8)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
4.5 C2H8N2 species
No insaturation is possible with the C2H8N2 formula, as illustrated in Table 5E. The most stable species is ethane-(1,1)-diamine, followed by ethane-(1,2)-diamine (called ethylenediamine), which lies ~6 kcal mol−1 above. The energy gap is comparable with that found between the second isomer (ethene-(1,1)-diamine) and the fourth (Z)-ethene-(1,2)-diamine in the preceding C2H6N2 series (Table 5D), namely of approximately 7 kcal mol−1. According to previous conformational studies and the microwave spectrum (Markov & Møllendal 1978), the most stable gauche conformation of ethylenediamine is considered here, with dipole moment of around 2.2 Debye.
The third on the energy scale, N-methyl methanediamine, is less stable than the ethane-(1,2)-diamine. The second and third isomers show energy differences of ~3 kcal mol−1 and ~9 kcal mol−1 respectively higher than the most stable isomer, ethane-(1,1)-diamine. With a dipole moment around 1.5 Debye, this N-methyl methanediamine includes the more stable sequence [NCN]. At more than 25 kcal mol−1 from the first isomer, the last compounds are hydrazines (ethyl-, (1,1)-dimethyl- and (1,2)-dimethyl-), containing the more brittle chain [CNN].
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C3H2N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
5 From C3H2N2 to C3H4N2
5.1 C3H2N2 species
We selected eight compounds with the formula C3H2N2 (Table 6). The first, malononitrile, is thermodynamically much more stable than any of the following isomers on this energy scale (about 12 kcal/mol for the second and 22 kcal/mol for the third and the fourth). Based on the MEP, we hypothesize that the first one, NC−CH2−CN, should be easily detected with its dipole moment of almost 4 Debye.
The case of C3H2N2 appears to be an anomaly, because even the most stable compound, the malononitrile NC−CH2−CN, has never been detected, even after the recent publication of its millimetric spectrum (Motiyenko et al. 2019). Millimetric spectra of the mono- (NC−CH2−NC, 2-isocyanoacetonitrile) and bis-isocyanide (CN−CH2−NC, diisocyanomethane) (Motiyenko et al. 2012) have been recorded, as has the microwave spectrum of 3-aminopropynenitrile (H2N−C≡C−CN) at the third position on the energy scale in Table 6 (Kolesniková et al. 2021). However, none of these species have been identified in the ISM so far.
5.2 C3H4N2 species
We investigated three different connectivities for the five heavy atoms: five-membered rings, open chains, and substituted three-membered rings. The microwave or millimetric spectrum of the majority of the most stable isomers have been reported: 1H-imidazole (Giuliano et al. 2019), (Z)-3-amino-2-propenenitrile (Askeland et al. 2006; Alberton et al. 2023), 1H-pyrazole (Kirchhoff 1967; Wlodarczak et al. 1991), 3-iminopropanenitrile (Lukova, Kolesnikova et al. 2022), and 2-aminopropenenitrile (Bauder & Ha 1983). To date, none of the C3H4N2 isomers have been identified in the ISM.
The results reported in Table 7 for the 23 low-lying isomers show that 1H-imidazole, stabilized by its aromaticity, is unambiguously the most stable species6. Its dipole moment close to ~4 Debye makes it a most reasonable target for observations.
The second compound on the energy scale is the conjugated (Z)-3-amino-2-propenenitrile called also (Z)-3-aminoacrylonitrile, H2N−CH=CH−CN. It lies ~5 kcal mol−1 above the first isomer. The value of its dipole moment, which is around 5 Debye, is encouraging for radio detection. It is followed by its diastereomer (E)-3-amino-2-propenenitrile, which is less stable, with a relative energy of almost 2 kcal mol−1 above, in agreement with previous works (Benidar et al. 2005). The aromatic pyrazole is the fourth most stable compound of the series, with a relative energy of around 10 kcal mol−1 above the aromatic imidazole and a lower dipole moment of ~2.3 Debye. There are four nitriles ranked between the positions 5 and 8 on this energy scale (Table 7). These are still conjugated but are probably highly electrophilic, as in 2-aminopropenenitrile or (E)-3-iminopropanenitrile (Benidar et al. 2005).
The series of cyanides follows at ~15 kcal mol−1 above the most stable isomer imidazole, as methyleneaminoacetonitrile (Motiyenko et al. 2013) at thirteenth position. Generally, isocyanides are placed on the energy scale with a difference around 20 kcal mol−1 higher than their corresponding nitriles. These isocyanides are ~30 kcal mol−1 above the 1H-imidazole, as (E)-2-isocyanoethene-1-amine or 1-isocyanoethene-1-amine at 15th and 16th positions. Between these cyanides in positions 9 and 10, nonaromatic five-membered ring isomers, 4H- and 2H-imidazole, come at 17 kcal mol−1 above their aromatic analog (1H), the most stable isomer of the series. Pyrazole analogs (4H, 3H) are higher on the energy scale, that is around 33 kcal mol−1 higher than 1H-imidazole.
Such an energy difference does not rule out the possibility of detecting these species as demonstrated by the identifications of H3C–NC (Gratier et al. 2013) and H-C≡C-NC (Kawaguchi, Ohishi et al. 1992), which are ~24 and 27 kcal mol−1 above H3C–CN (Solomon et al. 1971) and H-C≡C-CN (Lattelais et al. 2009), respectively. The chiral cyanoaziridine (2-cyanoaziridine) (Brown et al. 1980) is high in energy, and is the 17th isomer on the energy scale, at more than 33 kcal mol−1 above the most stable, 1H-imidazole.
Relative energy scale ΔE+ZPE (kcal mol−1) of C3H4N2 molecules using B3LYP/cc-pVQZ (B2) level of theory.
6 Conclusions
The aim of this study is to obtain information about the possibility of detecting new compounds containing two nitrogen atoms in species of increasing dimension from three to nine atoms, with these latter containing one more atom than the largest species identified in the ISM. This work was developed with a similar methodological approach to previous studies on five-membered heteroaromatic rings (Lattelais et al. 2010a). For these rings containing four carbons, one heteroatom (O, N, S), and a sufficient number of hydrogen atoms to preserve a six-electron aromatic shell, the aromaticity is a well admitted criterion of molecular stability. Using density functional theory, these previous studies found that pyrrole, furan, and thiophene are unambiguously the most stable isomers at the 10–50 K temperatures of the cold molecular clouds of the ISM.
In this work, we look at the problem of finding new molecules in space from another angle, in that we focus not on a global property such as aromaticity but on the possible presence of a local atomic sequence composed of two nitrogen atoms plus one carbon (2N + 1C) in either form [NCN or CNN]. Regarding C4H5N, C4H4O and C4H4S families, we also evaluated the thermodynamic stabilities with the help of DFT. To make sure that these DFT results are reliable, we designed an approach starting from CN2 up to C3H4N2, progressively increasing the size of the target molecule CxHyNz with x=(1,2,3), y=(0,2,4,6,8), keeping z=2 in all species and progressively increasing the level of theory up to coupled cluster whenever possible, that is, while the calculations remained reasonably tractable. For the smaller molecules, the panel of computations first employs DFT in the well-established B3LYP functional using 6-311++G** referred to as (B1), and then the correlation consistent basis cc-pVQZ (B2). The latter basis set, which is more adapted to the treatment of the electronic correlation, is used in all subsequent coupled-cluster treatments. The chaining of the calculations by increasing quality levels is DFT(B1) < DFT(B2) < CCSD(B2)/DFT(B2) < CCSD(B2). This progressive approach is applied until the chemical size of the target molecule makes the computational size of the system prohibitive. In this case, beyond C3H2N2, only DFT(B2) calculations were performed systematically. Therefore, here, new bis-nitrogen-containing functional groups are investigated, such as urea and methylisocyanide, confirming that correct energy scales can be obtained most of the time at the DFT level associated with the large basis set (B2). By applying this strategy to the entirety of the considered series, the linked sequence [NCN] appears more stable than the sequence [CNN].
The first important result is that the necessary preliminary checks (Table 1), show that the 14 bis-nitrogen species already identified in the ISM comply with the MEP. Therefore, from the 15 most relevant detected molecules collected in Table 8, our conclusions are as follows:
- (i)
With one carbon, the most stable molecules, namely those susceptible to identification7, are the couple of diastereomers of the formamidine (Z)- and (E)-H2N−CH=NH, methanediamine H2N−CH2−NH2 and, possibly also the NCN triplet in high magnetic fields. Clearly, the NCN isomer is always more stable than the CNN.
Table 8Most probable targets for future observations.
- (ii)
With two carbons, one can focus on a larger number of molecules whose detection could be possible, that is to say, HN=C=N−CH3, NC−NH−CH3, (Z)- and (E)-HN=CH−NH−CH3, (H2N)2C=CH2, and (H2N)2CH−CH3.
- (iii)
With three carbons, the most stable species awaiting detection are the heteroaromatic ring C3H4N2, imidazole, which comes before the diastereomers 3-aminoacrylonitrile, (E)- and (Z)-H2N−CH=CH−CN.
As predicted among the most probable targets to be observed, N-cyanomethanimine († in Table 8) has been effectively detected by San Andrés et al. (2024). Based on Table 8, Figure 1 gathers structures of the 15 most stable, potentially detectable candidates for future observations.
The theoretical dipole moment and rotational constants of each probable target isomer can be compared with the corresponding experimental data in Table B.1. Calculated values are in good agreement with experimental published data with an average uncertainty on the order of a few percent.
Finally, we identified a thermodynamically favored 3-atomic sequence namely [NCN] that is similar to the [NCO] sequence (Fourré et al. 2020). This study confirms that, within the limits of thermodynamics, the MEP is capable of accounting for the presence of all the bis-nitrogen species detected so far. It is an efficient and easily applicable tool for determining which isomers in a given series have the highest probability of being detected (within the strong constraints of a sufficient large dipole moment). When looking for bis-nitrogen species those containing the [NCN] sequence should be prioritized over [CNN] for further laboratory studies in preparation for next observation campaigns.
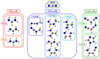 |
Fig. 1 Structures of the 15 most stable and probable target CxHyN2 molecules by isomer family for future observations. For energy ΔEZPE, dipole moments μ and rotational constants (A, B, C), see Table 8. |
Acknowledgements
This work was supported by CNRS national program PCMI (Physics and Chemistry of the Interstellar Medium) and by ENSC-Rennes.
Appendix A Nitrogen containing molecules
The following table A.1 gives a list of all the nitrogen containing species whose detection has been published as of today according to our knowledge of the literature.
Organic molecules detected in space so far containing Nitrogen atoms as function of the total number of atoms (those with two Nitrogen atoms are in bold characters).
Appendix B Dipole moments and experimental rotational constants
The following table B.1 gives common dipole moments and experimental rotational constants of CxHyN2 isomers by series when published to date according to our knowledge of the literature.
Experimental dipole moments and rotational constants (A, B, C) for isomers among most probable targets for future observations. Dipole moments μ are in Debye and rotational constants in GHz.
Appendix C About the role of diffuse atomic functions
The following table C.1 gathers relative ΔE+ZPE energies and dipole moments for selected model molecules using different basis sets: (B0) 6-311G**, (B1) 6-311++G**, (B2) cc-pVQZ, (B3) aug-cc-pVQZ. All relative energies are given at the DFT level of theory with the selected basis set.
Relative energy ΔE+ZPE (kcal mol−1) of selected isomers by series (A, B, C, D) using different basis sets with including or not diffuse atomic functions: (B0, B1, B2, B3). Dipole moments μ are in Debye.
References
- Agúndez, M., Cernicharo, J., de Vicente, P., et al. 2015, A&A, 579, A10 [Google Scholar]
- Agúndez, M., Marcelino, N., & Cernicharo, J. 2018, ApJ, 861, A22 [Google Scholar]
- Agúndez, M., Cabezas, C., Marcelino, N., et al. 2023, A&A, 669, A1 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Alberton, D., Lattanzi, V., Endres, C., et al. 2023, ApJ, 951, 108 [NASA ADS] [CrossRef] [Google Scholar]
- Askeland, E., Møllendal, H., Guillemin, J.-C., et al. 2006, J. Phys. Chem. A, 110, 12572 [NASA ADS] [CrossRef] [Google Scholar]
- Bak, B., & Svanholt, H. 1980a, Acta Chem. Scand., A34, 57 [CrossRef] [Google Scholar]
- Bak, B., & Svanholt, H. 1980b, Chem. Phys. Lett., 75, 528 [NASA ADS] [CrossRef] [Google Scholar]
- Bartlett, R. J., & Shavitt, I. 1977, Int. J. Quantum Chem., 12, 165 [Google Scholar]
- Bauder, A., & Ha, T.-K. 1983, Chem. Phys. Lett., 97, 135 [NASA ADS] [CrossRef] [Google Scholar]
- Becke, A. D. 1993, J. Chem. Phys., 98, 5648 [Google Scholar]
- Belloche, A., Menten, K. M., Comito, C., et al. 2008, A&A, 482, 179 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Belloche, A., Garrod, R. T., Müller, H. S. P., et al. 2019, A&A, 628, A10 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Benidar, A., Guillemin, J.-C., Otilia, Mó, & Yáñez, M. 2005, J. Phys. Chem. A, 109, 4705 [NASA ADS] [CrossRef] [Google Scholar]
- Bera, P. P., Noneman, K. K., & Lee, T. J. 2022, J. Phys. Chem. A, 126, 4700 [NASA ADS] [CrossRef] [Google Scholar]
- Bermúdez, C., Tercero, B., Motiyenko, R. A., et al. 2018, A&A, 619, A92 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Brown, R. D., Godfrey, P. D., & Ottrey, A. L. 1980, J. Mol. Spectr., 82, 73 [CrossRef] [Google Scholar]
- Evans, R. A., Lorencak, P., Ha, T.-K., & Wentrup, C. 1991, J. Am. Chem. Soc., 113, 7261 [CrossRef] [Google Scholar]
- Fourré, I., Rosset, L., Chevreau, H., & Ellinger, Y. 2016, A&A, 589, A18 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fourré, I., Matz, O., Pauzat, F., Ellinger, Y., & Guillemin, J.-C. 2020, A&A, 639, A16 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fraser, G. T., Leopold, K. R., Nelson, D. D., Tung, Jr., A., & Klemperer, W. 1984, J. Chem. Phys., 80, 3073 [NASA ADS] [CrossRef] [Google Scholar]
- Frisch, M., Trucks, G., Schlegel, B., et al. 2016, Gaussian 09, Revision A.02 and Gaussian 16, Revision C.01 (Wallingford, CT: Gaussian Inc.) [Google Scholar]
- Giuliano, B. M., Bizzocchi, L., Charmet, A., et al. 2019, A&A, 628, A53 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Gratier, P., Pety, J., Guzmán, V., et al. 2013, A&A, 557, A101 [CrossRef] [EDP Sciences] [Google Scholar]
- Karton, A., & Talbi, T.-K. 2014, Chem. Phys., 436, 22 [Google Scholar]
- Kawaguchi, K., Ohishi, M., Ishikawa, S-I., & Kaifu, N. 2009, ApJ, 386, L51 [Google Scholar]
- Kirchhoff, A. D. 1967, J. Am. Chem. Soc., 89, 1312 [CrossRef] [Google Scholar]
- Knauth, D. C., Andersson, B.-G., McCandliss, S. R., & Moos, H. W. 2004, Nature 409, 636 [NASA ADS] [CrossRef] [Google Scholar]
- Koch, W., & Holthausen, M. C. 2001, A Chemist Guide to Density Functional Theory, 2nd edn. (Weinheim, Germany: Wiley-VCH) [Google Scholar]
- Kolesniková, L., León, I., Alonso, E. R., Mata, S., & Alonso, J. L. 2021, Angew. Chem. Int. Ed., 60, 24461 [CrossRef] [Google Scholar]
- Lattelais, M., Pauzat, F., Ellinger, Y., & Ceccarelli, C. 2009, ApJ, 696, L133 [Google Scholar]
- Lattelais, M., Ellinger, Y., Matrane, A., & Guillemin, J.-C. 2010a, Phys. Chem. Chem. Phys., 12, 4165 [Google Scholar]
- Lattelais, M., Pauzat, F., Ellinger, Y., & Ceccarelli, C. 2010b, A&A, 519, A30 [CrossRef] [EDP Sciences] [Google Scholar]
- Lee, T. J., & Scuseria, G. E. 1995, in Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy, ed. S. R. Langhoff (Dordrecht, The Netherlands: Kluwer Academic Publishers) [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. 1988, Phys. Rev. B, 37, 785 [Google Scholar]
- Lukova, K., Kolesnikova, L., Koucky, J., et al. 2022, A&A, 665, A9 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Marstokk, K.-M., & Møllendal, H. 1978, J. Mol. Struct., 544, A82 [Google Scholar]
- McGuire, B. A., Loomis, R. A., Charness, C. M., et al. 2012, ApJ, 758, L33 [NASA ADS] [CrossRef] [Google Scholar]
- Merke, I., & Coudert, L. H. 2006, J. Mol. Spectr., 237, 174 [NASA ADS] [CrossRef] [Google Scholar]
- Motiyenko, R. A., Margules, L., & Guillemin, J.-C. 2012, A&A, 544, A82 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Motiyenko, R. A., Margules, L., & Guillemin, J.-C. 2013, A&A, 559, A44 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Motiyenko, R. A., Armieieva, I. A., Margulès, L., Alekseev, E. A., & Guillemin, J.-C. 2019, A&A, 623, A162 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pierce, L. Sr., & Dobyns, V. 1962, J. Am. Chem. Soc., 13, 2651 [CrossRef] [Google Scholar]
- Ramal-Olmedo, J. C., Menor-Salván, C. A., Miyoshi, A., & Fortenberry, R. C. 2023, A&A, 672, A49 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Raghachavari, K., Trucks, G. W., Pople, J. A., & Head-Gordon, M. 1989, Chem. Phys. Lett., 157, 479 [Google Scholar]
- Rivilla, V. M., Martín-Pintado, J., Jiménez-Serra, I., et al. 2019, MNRASL, 483, L114 [CrossRef] [Google Scholar]
- Rivilla, V. M., Jiménez-Serra, I., García de la Concepción, J., et al. 2021, MNRAS, 506, L79 [NASA ADS] [CrossRef] [Google Scholar]
- San Andrés, D., Rivilla, V. M., Colzi, L., Jiménez-Serra, I., et al. 2024, ApJ, 967, 39 [CrossRef] [Google Scholar]
- Schäfer, E., Winnewisser, M., & Christiansen, I. J. 1981, Chem. Phys. Lett., 81, 380 [CrossRef] [Google Scholar]
- Solomon, P. M., Jefferts, K. B., Penzias, A. A., & Wilson, R. W. 1971, ApJ, 168, L107 [Google Scholar]
- Steinmetz, W. 1970, J. Chem. Phys., 52, 2788 [NASA ADS] [CrossRef] [Google Scholar]
- Stephens, P. J., Devlin, F. J., Chabalowski, C. F., & Frisch, M. J. A. 1994, J. Phys. Chem., 98, 11623 [CrossRef] [Google Scholar]
- Szabo, A., & Ostlund, N. S. 1989, in Handbook Introduction to Advanced Eletronic Structure Theory, Modern Quantun Chemistry (New York: McGraw-Hill) [Google Scholar]
- Turner, B. E. 1974, ApJ, 193, L83 [Google Scholar]
- Turner, B. E., Liszt, H. S., Kaifu, N., & Kisliakov, A. G. 1975, ApJ, 201, L149T [NASA ADS] [CrossRef] [Google Scholar]
- Wasserman, E., Barash, L., & Yager, W. A. 1965, J. Am. Chem. Soc., 87, 2075 [CrossRef] [Google Scholar]
- Winnewisser, M., Winnewisser, B.P., & Wentrup, C. 1984, J. Mol. Spectr., 105, 193 [CrossRef] [Google Scholar]
- Wlodarczak, G., Demaison, J., van Eijck, B. P., Zhao, M., & Boggs, J. E. 1991, J. Chem. Phys., 94, 6698 [NASA ADS] [CrossRef] [Google Scholar]
- Zaleski, D. P., Seifert, N. A., Steber, A. L., et al. 2013, ApJ, 765, L10 [Google Scholar]
- Zhang, X., Quan, D., Chang, Q., et al. 2020, MNRAS, 497, 609 [NASA ADS] [CrossRef] [Google Scholar]
- Zhang, X., Quan, D., Li, R., et al. 2023, MNRAS, 521, 1578 [NASA ADS] [CrossRef] [Google Scholar]
- Ziurys, L. M., Apponi, A. J., Hollis, J. M., & Snyder, L. E. 1994, ApJ, 436, L181 [NASA ADS] [CrossRef] [Google Scholar]
The important class of protonated or zwitter-ionic species will be considered in an other context.
For a comprehensive presentation of DFT methods and their ability to reproduce the structural properties, energies and spectroscopic parameters (vibration frequencies, dipole moments, etc.) of organic molecules, we refer the reader to Lee & Scuseria (1995) and Koch & Holthausen (2001). Concerning multideterminental incorporation of electronic correlation into the wave function (coupled-cluster theories (CCSD/CCSDT)) (Bartlett & Shavitt 1977; Raghachavari et al. 1989), an easier insight into post-HF methods is provided in Szabo & Ostlund (1989).
Taken from DFT(B2) vibrational analysis.
The acronym CCSD/DFT(B2) means a single point CCSD calculation at the DFT(B2) optimized geometry which is relevant and computationally less expensive than CCSD(B2).
Except for 3-amino-2-propenenitrile, when an isomer have two configurations (E) and (Z), the two corresponding diastereomers have been computed, but only the most stable is shown in Table 7.
At this point, the H3N−HCN complex has not been retained among the preferred candidates in view of the variability of its ranking on the energy scale when increasing the level of correlation. Further studies are necessary to clarify this point unambiguously.
All Tables
Molecules detected in space so far containing two nitrogen atoms as a function of the total number of atoms (the chronological order of detection is shown in parenthesis).
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing CH(y=0,2,4,6)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=0, 2)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=4)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C2H(y=6,8)N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative ΔE+ZPE energies (kcal mol−1) of molecules containing C3H2N2 atoms using 6-311++G** (B1) and cc-pVQZ (B2) basis sets.
Relative energy scale ΔE+ZPE (kcal mol−1) of C3H4N2 molecules using B3LYP/cc-pVQZ (B2) level of theory.
Organic molecules detected in space so far containing Nitrogen atoms as function of the total number of atoms (those with two Nitrogen atoms are in bold characters).
Experimental dipole moments and rotational constants (A, B, C) for isomers among most probable targets for future observations. Dipole moments μ are in Debye and rotational constants in GHz.
Relative energy ΔE+ZPE (kcal mol−1) of selected isomers by series (A, B, C, D) using different basis sets with including or not diffuse atomic functions: (B0, B1, B2, B3). Dipole moments μ are in Debye.
All Figures
|
Fig. 1 Structures of the 15 most stable and probable target CxHyN2 molecules by isomer family for future observations. For energy ΔEZPE, dipole moments μ and rotational constants (A, B, C), see Table 8. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.