A&A 491, 907-916 (2008)
DOI: 10.1051/0004-6361:200810374
Photodesorption of water ice
A molecular dynamics study
S. Andersson1,2,3 - E. F. van Dishoeck1
1 - Leiden Observatory, Leiden University, PO Box 9513, 2300
RA Leiden, The Netherlands
2 -
Gorlaeus Laboratories, Leiden Institute of Chemistry,
Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands
3 -
Department of Chemistry, Physical Chemistry, Universtity of Gothenburg,
41296 Gothenburg, Sweden
Received 11 June 2008 / Accepted 24 September 2008
Abstract
Context. Absorption of ultraviolet radiation by water ice coating interstellar grains can lead to dissociation and desorption of the ice molecules. These processes are thought to be important in the gas-grain chemistry in molecular clouds and protoplanetary disks, but very few quantitative studies exist.
Aims. We compute the photodesorption efficiencies of amorphous water ice and elucidate the mechanisms by which desorption occurs.
Methods. Classical molecular dynamics calculations were performed for a compact amorphous ice surface at 10 K thought to be representative of interstellar ice. Dissociation and desorption of H2O molecules in the top six monolayers are considered following absorption into the first excited electronic state with photons in the 1300-1500 Å range. The trajectories of the H and OH photofragments are followed until they escape or become trapped in the ice.
Results. The probability for H2O desorption per absorbed UV photon is 0.5-1% in the top three monolayers, then decreases to 0.03% in the next two monolayers, and is negligible deeper into the ice. The main H2O removal mechanism in the top two monolayers is through separate desorption of H and OH fragments. Removal of H2O molecules from the ice, either as H2O itself or its products, has a total probability of 2-3% per absorbed UV photon in the top two monolayers. In the third monolayer the probability is about 1% and deeper into the ice the probability of photodesorption falling to insignificant numbers. The probability of any removal of H2O per incident photon is estimated to be
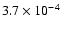 ,
with the probability for photodesorption of intact H2O molecules being
,
with the probability for photodesorption of intact H2O molecules being
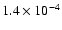 per incident photon. When no desorption occurs, the H and OH products can travel up to 70 and 60 Å inside or on top of the surface, respectively, during which they can react with other species, such as CO, before they become trapped.
per incident photon. When no desorption occurs, the H and OH products can travel up to 70 and 60 Å inside or on top of the surface, respectively, during which they can react with other species, such as CO, before they become trapped.
Key words: astrochemistry - molecular data - ISM: molecules
Ices are a major reservoir of the heavy elements in a variety of
astrophysical environments, ranging from cold and dense molecular
clouds (e.g., Whittet et al. 1988; Pontoppidan et al. 2004; Willner et al. 1982; Murakawa et al. 2000; Smith et al. 1989) and protoplanetary disks
(Pontoppidan et al. 2005; Terada et al. 2007)
to the icy bodies in our own solar system such as comets
(e.g., Mumma et al. 1993) and Kuiper Belt Objects
(Jewitt & Luu 2004). In star-forming
clouds, the fraction of carbon and oxygen locked up in ice is
comparable to that in gaseous CO (Pontoppidan 2006; van Dishoeck et al. 1996),
whereas at the centers of cold pre-stellar cores more than 90% of the
heavy elements can be frozen out (e.g., Bergin et al. 2002; Caselli et al. 1999).
Similarly, the cold midplanes of protoplanetary disks
around young stars are largely devoid of gaseous molecules other than
H2, H3+ and their isotopologues (e.g., Aikawa et al. 2002; Ceccarelli & Dominik 2005).
Thus, a good understanding of how molecules adsorb
and desorb from the grains is critical to describe the chemistry in
regions in which stars and planets are forming.
The importance of ultraviolet (UV) radiation in affecting interstellar
ices is heavily debated in the literature. On the one hand, the large
extinctions of 100 mag or more along the lines of sight where ices are
detected prevent UV radiation from penetrating deep into the clouds (e.g., Stäuber et al. 2004; Ehrenfreund et al. 2001), unless there are
cavities through which the stellar UV photons can escape (Spaans et al. 1995). Thus,
the bulk of the ices are thought to be shielded from both external and
internal radiation sources in which case photodesorption is thought to be
unimportant (e.g., Hartquist & Williams 1990; Léger et al. 1985). On the other
hand, UV photons are produced locally throughout the cloud by the
interaction of cosmic rays with the gas, albeit at a level about
104 times less than that of the general interstellar radiation
field (Prasad & Tarafdar 1983; Shen et al. 2004). In addition,
X-rays from young stars penetrate much further into their surroundings
than UV and can produce local UV photons through a similar process
(Stäuber et al. 2005; Dalgarno et al. 1999). Moreover, the
observed emission of optically thick millimeter lines from gaseous
molecules is often dominated by the outer layers of the cloud where UV
photons play a role. These UV photons can be important not only in
the desorption of ices but also in the creation of reactive
photo-products such as energetic H atoms and radicals which can move
through the ice and encounter other species leading to the formation
of more complex molecules (e.g., d'Hendecourt et al. 1982; Garrod & Herbst 2006).
Water ice is the dominant consituent of interstellar ices (e.g., Pontoppidan 2006; Gibb et al. 2000)
with an abundance at least three orders of magnitude larger than that
of gaseous water in cold clouds (Boonman et al. 2003; Snell et al. 2000). Thus,
evaporation of water ice, even at a low fraction, can significantly
affect the gaseous water abundance. Recent models of translucent and
dense clouds invoke photodesorption of water ice in the outer regions
to explain the gaseous water emission observed by the Submillimeter
Wave Astronomy Satellite (SWAS) (Bergin & Melnick 2005).
Photodesorption is also used to interpret the tentative
detections of HDO and other gaseous species in the surface layers of
protoplanetary disks (Willacy & Langer 2000; Dominik et al. 2005).
The adopted desorption efficiencies in these models, about 0.1% per
incident UV photon, are based on a single experiment by
Westley et al. (1995b,a) exposing ices at 35-100 K to Lyman-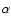 radiation.
The authors did not detect any water photodesorption in the limit of low UV
photon fluence (integrated flux). Therefore it was suggested that water photodesorption
only occurs from ices
that have been subject to large doses of UV photons and not directly upon the first
exposure to UV radiation. This is in contrast with recent experiments reported by Öberg
et al. (2008), where there is a clear component of the water photodesorbed from
amorphous ices at 18-100 K that
is detected directly upon the first exposure to a UV lamp. Apart from that the results
remain quite similar to the ones by Westley et al.
radiation.
The authors did not detect any water photodesorption in the limit of low UV
photon fluence (integrated flux). Therefore it was suggested that water photodesorption
only occurs from ices
that have been subject to large doses of UV photons and not directly upon the first
exposure to UV radiation. This is in contrast with recent experiments reported by Öberg
et al. (2008), where there is a clear component of the water photodesorbed from
amorphous ices at 18-100 K that
is detected directly upon the first exposure to a UV lamp. Apart from that the results
remain quite similar to the ones by Westley et al.
Other experiments on UV irradiation of water ices have also been performed.
Ghormley & Hochanadel (1971) observed H, OH, and H2O2 following UV irradiation
of crystalline ice at 263 K, while Gerakines et al. (1996) found production of OH, HO2, and
H2O2 in the ice upon exposing amorphous ice at 10 K to UV light covering
mainly the first and second electronic absorption bands of H2O. Watanabe et al. (2000)
irradiated amorphous D2O ice at 12 K with UV photons
and observed substantial amounts of D2 after
irradiation at
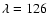 nm, but very little at
nm, but very little at
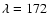 nm.
In the experiments by Yabushita et al. (2006) H atoms were found to desorb from the ice
after UV irradiation at
nm.
In the experiments by Yabushita et al. (2006) H atoms were found to desorb from the ice
after UV irradiation at
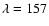 nm and
nm and
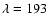 nm.
There are also a few reports on two- and multi-photon excitation of water ice leading
to photodesorption of H2O molecules (Bergeld & Chakarov 2006; Nishi et al. 1984). In these cases
the photon energies are below the threshold for absorption in the ice, but
upon multiple absorptions the excitation energies fall between 9 and 10 eV, between
the first and second absorption band in ice.
Clearly, there is a need for more quantitative information
on the processes induced by UV photons in ices, even for the simplest
cases such as pure water ice.
nm.
There are also a few reports on two- and multi-photon excitation of water ice leading
to photodesorption of H2O molecules (Bergeld & Chakarov 2006; Nishi et al. 1984). In these cases
the photon energies are below the threshold for absorption in the ice, but
upon multiple absorptions the excitation energies fall between 9 and 10 eV, between
the first and second absorption band in ice.
Clearly, there is a need for more quantitative information
on the processes induced by UV photons in ices, even for the simplest
cases such as pure water ice.
We present here the results of the first theoretical study of the
dissociation of H2O molecules in pure water ice following
absorption by UV photons. In addition to providing probabilities for
desorption to be used in astrochemical models, these simulations
provide insight into the mechanisms leading to desorption as well as
the movements of the energetic photoproducts in the ice before they
become trapped. In Sect. 2, we present the methods used in this
study, in Sect. 3 the main results, and in Sect. 4 a short
discussion and astrochemical implications. In Sect. 5 the results are summarized
and some concluding remarks are given.
All our calculations have been performed using classical Molecular
Dynamics (MD) methods (Allen & Tildesley 1987) with analytical
potentials. Details of the computational procedure have been described
in Andersson et al. (2006); here only a brief outline of
the methods will be presented.
To create an amorphous ice slab, the procedure outlined in Al-Halabi et al. (2004a)
was used. In brief, a slab of 8 bilayers (16 monolayers) of crystalline ice was first created consisting of a cell
containing 480 H2O molecules. The cell has the dimensions x: 22.4 Å, y: 23.5 Å, and z: 29.3 Å. Periodic boundary
conditions are applied in the x- and y-directions, the z coordinate being parallel to the surface normal. Thus an infinite ice
surface is created. The H2O molecules are treated as rigid rotors
and their interactions are governed by the TIP4P potential (Jorgensen et al. 1983),
which describes the interaction as a sum
of pair interactions (electrostatic and Lennard-Jones potentials). The
two bottom bilayers are kept fixed to simulate bulk ice and the
molecules in the other 6 bilayers are allowed to move without any
dynamical constraints other than that they remain rigid. The dynamics
are at all times governed by classical Newtonian mechanics. To force
the transition to amorphous ice, the surface is initially allowed to
equilibrate for 5 ps at 10 K but then the temperature is increased to 300 K using a computational equivalent of a thermostat (Berendsen et al. 1984).
In this way the top bilayers form a
liquid. The system is left to equilibrate for 100 ps after which it is
rapidly cooled to 10 K. Then it is once again equilibrated for 100 ps. The resulting amorphous ice structure most closely resembles the
structure of compact amorphous ice obtained
experimentally and is thought to be representative of the
structure of interstellar water ice (Al-Halabi et al. 2004b,a).
See Fig. 4 of Al-Halabi et al. (2004a) and Fig. 2 of Andersson et al. (2006) for images.
It does
not exhibit the microporous structure that is obtained in vapor
deposited ice (Mayer & Pletzer 1986; Kimmel et al. 2001b,a).
Given the dimensions of the simulation cell
such a structure is simply not possible to obtain, since the pores should have a size on
the same order as the cell we use.
However, we believe that it
is a good representation of an amorphous ice surface on a local scale, whether that
be at the ``outside'' of the surface or inside a void deeper in the ice.
Implications of this for the obtained results are discussed in Sect. 4.
In the rest of the paper we will discuss the depth into the ice in terms
of monolayers. To avoid confusion our definition of monolayer is
the thickness of ice corresponding to half a crystalline bilayer, i.e., if
the ice were crystalline each bilayer would consist of two monolayers.
For ease of definition the monolayers have been taken to be divided according
to the z values of the centers-of-mass of the molecules, e.g., the 30 molecules
with the largest values of z constitute the top (``first'') monolayer.
Once the ice surface is set up, one H2O molecule is chosen to be
photodissociated. This molecule is then made completely flexible and
its intramolecular (internal) interactions are governed by an analytic
potential energy surface (PES) for the first electronically excited state
(the Ã
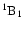 state) of
gas-phase H2O based on high-quality ab initio electronic
structure calculations (Dobbyn & Knowles 1997). This excited
potential is fully repulsive so that absorption into this state leads
to dissociation of the H2O molecule into H + OH. The intermolecular
interactions of the excited state H2O with the surrounding H2O
molecules are governed by specially devised partial charges for
describing the electrostatic interactions.
In short, a charge of -0.2e is put on the O atom and charges of
+0.1e on the H atoms. This gives a smaller dipole moment than
that of the ground state H2O potential. The effect of this is
that a less favorable interaction is obtained with the surrounding H2O
molecules, giving higher excitation energies than with the uncorrected
gas-phase potential energy surface. This leads to the blue-shift of about 1 eV of
the ice UV spectra seen in Fig. 1, which
agree well with the first UV absorption band in amorphous and
crystalline ices. If the ground state partial charges
were to be used for the excited state the excitation spectrum would
coincide with the gas-phase UV spectrum.
For more
details on the potentials see Andersson et al. (2006).
Similar procedures using potential energy surfaces for higher
excited states (B, C, etc.) should in principle give reasonable
representations of higher-lying absorption bands in water ice.
state) of
gas-phase H2O based on high-quality ab initio electronic
structure calculations (Dobbyn & Knowles 1997). This excited
potential is fully repulsive so that absorption into this state leads
to dissociation of the H2O molecule into H + OH. The intermolecular
interactions of the excited state H2O with the surrounding H2O
molecules are governed by specially devised partial charges for
describing the electrostatic interactions.
In short, a charge of -0.2e is put on the O atom and charges of
+0.1e on the H atoms. This gives a smaller dipole moment than
that of the ground state H2O potential. The effect of this is
that a less favorable interaction is obtained with the surrounding H2O
molecules, giving higher excitation energies than with the uncorrected
gas-phase potential energy surface. This leads to the blue-shift of about 1 eV of
the ice UV spectra seen in Fig. 1, which
agree well with the first UV absorption band in amorphous and
crystalline ices. If the ground state partial charges
were to be used for the excited state the excitation spectrum would
coincide with the gas-phase UV spectrum.
For more
details on the potentials see Andersson et al. (2006).
Similar procedures using potential energy surfaces for higher
excited states (B, C, etc.) should in principle give reasonable
representations of higher-lying absorption bands in water ice.
![\begin{figure}
\par\includegraphics[angle=270,width=8cm,clip]{0374fig1.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg12.gif) |
Figure 1:
Calculated and experimental (Kobayashi 1983) spectra of
the first UV absorption band in crystalline and amorphous ices and
calculated first absorption band for gas-phase H2O. Note that
the intensity is given in arbitrary units and that the intensities
of the ice spectra have been scaled to roughly coincide. |
| Open with DEXTER |
The initial internal
coordinates and momenta of the atoms in the selected molecule are
sampled by a Monte Carlo procedure using a semi-classical (Wigner)
phase-space distribution (Schinke 1993) that has been
fitted to the ground-state vibrational wave function of H2O (van Harrevelt et al. 2001). This procedure gives initial
conditions that are very similar to those found in fully quantum
mechanical methods and has been shown to work well for the description
of photodissociation processes of gaseous molecules. The transition
dipole moment function, which governs the strength of the absorption,
is taken from the calculation for gaseous H2O by van Harrevelt & van Hemert (2000).
Dissociation of molecules in the top six monolayers has been
considered. For each monolayer all 30 molecules have been
dissociated, one molecule at a time.
For each molecule 200 configurations and momenta were
sampled from the Wigner distribution. This gives 6000 trajectories per
monolayer and 36 000 trajectories in total.
The excitation energy is computed by taking the energy difference
between an ice slab with an excited state H2O and one with a ground
state H2O (with the same coordinates). Each excitation is assigned
a weight calculated as the square of the coordinate-dependent
transition moment. By summing the weights of the excitation energies
binned in 0.05 eV-wide energy intervals, ``intensities'' are
obtained. Taken together these intensities form a UV absorption
spectrum for the ice. The monolayers 5 to 6 were found to be
converged to a ``bulk behavior'' (Andersson et al. 2005)
and could therefore be used to compare the calculated spectra to
experimental data. The gas-phase spectrum presented in Sect. 3 was obtained using the same intramolecular potential
surfaces as above, but without the surrounding molecules and with 1000 sampled configurations.
After putting the molecule in the excited state, the dissociating
trajectory is integrated with a timestep of 0.02 fs. A maximum time of 20 ps has been used before terminating the trajectory. Most of the
trajectories (99.6%) were terminated before that because the system was found
in one of the final outcomes (see Sect. 3) with
negligible probability of transforming into a different state.
When the excited H2O dissociates, the intermolecular interactions
are smoothly switched into separate interactions between the
photoproducts and water ice, i.e., H-H2O and OH-H2O
potentials. All details of the potentials and the functions used to switch between
different potentials are given in Andersson et al. (2006). The switching functions connect
the partial charges, the dispersion interactions and repulsive potentials between
the H2O-H2O potentials and the OH-H2O and H-H2O potentials affecting the
dissociating molecule. The switches are functions of the OH distances (
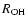 )
within this molecule
and will give the interaction parameters as continuous functions in the range 1.1-1.6 Å in
.
The intramolecular potential is switched to the
ground-state PES, which allows H and OH to recombine to form H2O. This
switch is done in an analogous way as for the intermolecular interactions above, but here the
range of
where the switch is made is 3.0-3.5 Å. In this range
the excited-state and ground-state PES are near-degenerate, so a high transition probability
between the two states is quite probable. Once
becomes larger than 3.5 Å the system will remain
on the ground-state PES, even if
again becomes smaller than 3.5 Å. This is what allows for recombination
of H and OH.
The intermolecular interactions for the recombined ground state H2O with the
surrounding H2O molecules are taken from the TIP3P potential
(Jorgensen et al. 1983).
)
within this molecule
and will give the interaction parameters as continuous functions in the range 1.1-1.6 Å in
.
The intramolecular potential is switched to the
ground-state PES, which allows H and OH to recombine to form H2O. This
switch is done in an analogous way as for the intermolecular interactions above, but here the
range of
where the switch is made is 3.0-3.5 Å. In this range
the excited-state and ground-state PES are near-degenerate, so a high transition probability
between the two states is quite probable. Once
becomes larger than 3.5 Å the system will remain
on the ground-state PES, even if
again becomes smaller than 3.5 Å. This is what allows for recombination
of H and OH.
The intermolecular interactions for the recombined ground state H2O with the
surrounding H2O molecules are taken from the TIP3P potential
(Jorgensen et al. 1983).
A slightly different stop criterion has been used in these calculations compared to the results presented
previously (Andersson et al. 2006,2005). When an H atom or OH is accommodated to the
ice surface (``trapped'') the trajectory is run until its translational energy equals
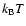 or lower
and the binding energy to the surface is 0.02 eV (H atom) or 0.1 eV (OH) or stronger. In the older
version of the code, the stop criterion was based on the kinetic energy of the individual atoms
in relation to the potential energy (Andersson et al. 2006). The introduction of the new termination
scheme led to a reduction by about 50% of the number of trajectories exceeding 20 ps.
or lower
and the binding energy to the surface is 0.02 eV (H atom) or 0.1 eV (OH) or stronger. In the older
version of the code, the stop criterion was based on the kinetic energy of the individual atoms
in relation to the potential energy (Andersson et al. 2006). The introduction of the new termination
scheme led to a reduction by about 50% of the number of trajectories exceeding 20 ps.
Although most of the results presented here focus on amorphous ice,
calculations have been performed for crystalline ice as well for
comparison. Details can be found in Andersson et al. (2006).
3 Results and discussion
As presented in Fig. 1, our calculated spectra of the
first UV absorption bands in amorphous and crystalline ice match very
well the experimentally obtained spectra, both in general shape as
well as in the peak and threshold energies. The calculated gas-phase
spectrum of the first absorption band shown in Fig. 1
also matches the experimental peak energy (7.4-7.5 eV) quite well.
This is naturally to be expected, since the potential surfaces used
are based on very high-quality ab initio calculations of the
energy. The success in reproducing the measured spectra leads us to
believe that the amount of excess energy released into the ice is
basically correct.
The ice spectrum is blue-shifted with respect to that of gaseous
H2O and has significant cross section only in the 7.5-9.5 eV
range. Thus, the photodesorption probabilities computed here are
appropriate for photons in the 1300-1500 Å range.
Dissociation of H2O can also occur following absorption into
higher excited states (e.g., the equivalent of the B state
of gaseous H2O) but these generally contribute less
than 20% of the total absorption in a dense cloud.
3.2 Photoprocesses and desorption probabilities
3.2.1 Overall probabilities
Photodissociation of a water ice molecule can have several outcomes,
with the H and OH photoproducts either becoming trapped in the ice,
recombining back to an H2O molecule, or desorbing from the ice
surface. Figure 2 shows the probabilities (as fractions
of the number of absorbed photons) of the main processes as functions
of how deep into the ice the dissociating molecule initially is located.
Note that these probabilities are
given per absorbed UV photons and not per incident photon.
In the first monolayer the dominant outcome is
that the hydrogen atom desorbs and the OH radical is
trapped in or on the ice. The probability of this event drops rapidly
from about 0.9 in the first monolayer to just over 0.1 in the sixth
monolayer. This reflects the effect of the ice on the motion of the H atom. At the surface there are very few molecules to stop the H atom
from desorbing, but starting from deeper into the ice there are more
obstacles on the way to the gas phase.
![\begin{figure}
\par\includegraphics[angle=270,width=8cm,clip]{0374fig2.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg15.gif) |
Figure 2:
Fractions of the main outcomes after H2O photodissociation for the top six monolayers of amorphous ice. These probabilities
are calculated from all trajectories, irrespective of excitation energy. |
| Open with DEXTER |
For the same reasons the probabilities of the other two major outcomes
steadily increase as one moves deeper into
the ice. These are the events when either both H and OH become separately trapped in the ice or when H and OH
recombine to form H2O to subsequently become trapped in the
ice. Except for the top two monolayers the probabilities of these two
events are roughly equal and in the sixth monolayer the probabilities
are up to 0.4. The reason for the lower probability of the H2O
molecule being recombined and trapped in the top monolayers can be
understood from the open structure of the uppermost layers, which more
easily allows for the photofragments to escape the region of the ice
where they were initially formed.
Table 1:
Total probabilities of H atom, OH, and H2O desorption (per absorbed UV photon) as functions of monolayer.
The probability of photodesorption of H2O is seen to be low
compared with the above processes, 0.7% in the top layer and 0.8% in the second layer, and
then decreases with distance from the surface (see Table 1).
However, this is only considering the desorption of intact H2O molecules.
If one is interested in the removal of H2O from the surface without considering what enters
the gas phase, the dominant mechanism in the top two monolayers is actually desorption
of separate H and OH fragments. Desorption of OH is in most cases accompanied by the
desorption of an H atom, but a minor fraction of OH desorption occurs with the H atom being
trapped in the ice (see Sect. 3.2.2).
In summary, the desorption probabilities of OH and H2O are about 2 orders of magnitude
lower than that of H atoms with OH desorption being about twice as probable as H2O desorption
if one sums over the probabilities from all monolayers (see also Sect. 4).
3.2.2 Mechanisms and their probabilities
Analysis of the
trajectories shows that there are three distinct mechanisms
for H2O removal (see Fig. 3). Note that these
snapshots are taken from calculations on crystalline ice
(Andersson et al. 2006) for ease of visualization: (a) An H atom
released from photodissociation of H2O is able to transfer enough
momentum to one of the other H2O molecules to ``kick'' it off the
surface, (b) H and OH recombine to form H2O and subsequently desorb, and (c) the H and OH both desorb from the surface separately.
In Table 2 the absolute probability of H2O desorption is given
along with the relative probabilities of the three different
mechanisms for each monolayer. With the additional mechanism (c) the
probability of H2O removal is 2.7% and 1.9% per absorbed UV photon in the
first and second monolayer, respectively.
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig3.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg21.gif) |
Figure 3:
Snapshots of trajectories of mechanisms of H2O
desorption for a crystalline ice model. a) One of the surrounding
molecules desorbs. b) The photofragments H and OH recombine and
desorb as H2O. c) The photofragments both desorb as separate
species. The red and white atoms correspond to O and H atoms in the
surrounding molecules and the blue and yellow atoms correspond to
the O and H atoms of the photodissociated H2O molecule. |
| Open with DEXTER |
In the top two monolayers the direct desorption of H and OH fragments is the dominant desorption
mechanism, but in the third layer the three distinct desorption mechanisms (``a'', ``b'', and ``c'') are
roughly equally probable.
Following UV absorption in monolayers 4 and 5 only the indirect ``kick-out''
mechanism is effective. In Fig. 4 the probabilities of all mechanisms of
desorption of H atoms, OH radicals, and H2O molecules are presented for the top five monolayers.
The desorption of OH is possible either together with the H atom as shown above or separately with
the H atom remaining trapped. The former mechanism is found to have a higher probability. In total,
the desorption of OH is about a factor of 2 more probable than the desorption of H2O. Below the
third monolayer the released OH radicals do not have sufficient kinetic energy to make it to the top of the
surface and desorb. The rightmost
three categories constitute a further division of the category of indirect H2O desorption.
Here ``H2O indirect desorption'' only refers to the cases where a molecule is kicked out by an H atom, which
subsequently remains in the ice. The category ``H2O desorption induced by recombination'' refers to the rare occurence
where it is the excess energy from the recombination of the H and OH fragments that kicks the molecule off the surface.
The case ``H + H2O desorb'' refers to when the H atom kicks the molecule off the surface and subsequently desorbs
itself. This last category dominates the indirect desorption in the first two monolayers and
also consitutes the maximum amount of matter that has been observed to desorb following
photoexcitation.
For H2O photoexcited below the fifth
monolayer there is no evidence of photodesorption of H2O molecules.
Table 2:
Absolute probabilities for removal of H2O from an amorphous ice
surface (per absorbed UV photon) as functions of monolayer.
![\begin{figure}
\par\includegraphics[angle=270,width=8.4cm,clip]{0374fig4.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg24.gif) |
Figure 4:
Fractions of the detailed outcomes after H2O
photodissociation for the top five monolayers of amorphous ice. The
error bars correspond to a 95% confidence interval. |
| Open with DEXTER |
The division into monolayers in Table 2 refers to where the
photoexcited H2O is situated.
The H2O molecules that actually desorb upon being expelled by an H atom or recombining H2O molecule
all originate in monolayers 1 (84%) and 2 (16%). In most of these cases it is not only the transfer
of momentum that is effective, but also the repulsive interaction from the photoexcited molecule,
which most often is in the near vicinity of the desorbing molecule. To illustrate this one can consider the lowering
in binding energy of the molecule about to be desorbed. In monolayer 1 the average binding energy of all molecules is 0.9 eV (calculated using the TIP4P potential) and of the desorbing molecules prior to excitation it is 0.8 eV.
However, the photoexcitation lowers the binding
energy of these molecules by on average 0.3 eV. About 25% of the desorbing molecules do not have their binding
energy significantly lowered by excitation, but are kicked out solely by momentum transfer.
If these molecules are excluded then the binding energy is on average lowered by 0.4 eV. In the second monolayer only about 10%
of the desorbing molecules do not have their binding energy significantly lowered. There the average binding energy
is 1.1 eV and this is lowered by 0.3 eV on average upon photoexcitation.
There is no sign of molecules being electronically excited and then desorbing intact directly, i.e.,
expelled by the repulsive interaction of the excited state molecule with its surroundings.
The molecules dissociate very quickly (on the order of 10 fs) and that is not sufficient time for the molecule to desorb
before it is dissociated. As discussed above the photofragments can however recombine and then
subsequently desorb as H2O.
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig5.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg25.gif) |
Figure 5:
The average initial translational energies of H atoms [
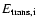 (H)] and OH radicals [
(OH)] and
the average OH vibrational energy following photodissociation as function of excitation energy
for the top six monolayers in amorphous ice. (H)] and OH radicals [
(OH)] and
the average OH vibrational energy following photodissociation as function of excitation energy
for the top six monolayers in amorphous ice. |
| Open with DEXTER |
3.2.3 Effects of product energies
The H atoms that are released have an average energy of 1.5-2.5 eV depending on the excitation energy
and to a lesser extent in which monolayer they originate (see Fig. 5). As can be seen the
average initial H atom translational energy increases with increasing excitation energy, but above an excitation
energy of 9 eV it drops to
somewhat lower energies. The average vibrational energy of OH has a minimum value of 0.3 eV around
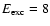 eV
but increases strongly to about 2 eV at
eV
but increases strongly to about 2 eV at
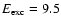 eV. This implies that at lower excitation energies
the vast majority of the
OH molecules are formed in the vibrational ground state, since the experimental zero-point energy of OH is 0.23 eV (Huber & Herzberg 1979) (see also Andersson et al. 2006). When the excitation energy is increased above 9 eV,
large fractions of vibrationally excited OH are produced.
The average initial translational energy of OH is only weakly
dependent on excitation energy and lies around 0.2 eV with only a slight increase with increasing
eV. This implies that at lower excitation energies
the vast majority of the
OH molecules are formed in the vibrational ground state, since the experimental zero-point energy of OH is 0.23 eV (Huber & Herzberg 1979) (see also Andersson et al. 2006). When the excitation energy is increased above 9 eV,
large fractions of vibrationally excited OH are produced.
The average initial translational energy of OH is only weakly
dependent on excitation energy and lies around 0.2 eV with only a slight increase with increasing
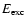 .
.
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig6.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg29.gif) |
Figure 6:
The probabilities of H atom desorption and trapping of H and OH as function of initial
H atom translational energy averaged over the top six monolayers in amorphous ice.
Note that for technical reasons these are probabilities with the possibility of H2O recombination
excluded (see text). The error bars correspond to a 95% confidence interval. |
| Open with DEXTER |
In Fig. 6 the H atom desorption probability is plotted alongside the probability of H and OH both becoming
trapped as functions of initial H atom translational energy.
The plotted probabilities are somewhat higher than they should be because
the probability of recombination of H2O has been excluded in the set of outcomes. The reason for this
is that during recombination the translational energy of the H atom becomes very high. If recombination occurs immediately
after dissociation it is quite difficult to distinguish the maximum translational energy the H atom normally would have
after photodissociation and the maximum translational energy it gets during recombination. However, the trend
is clear that the H atom desorption probability increases with increasing initial translational energy, as one would
intuitively expect. The reason for the unexpectedly high desorption probability at
(H) = 0.7 eV is not quite
clear and it could simply be an effect of insufficient sample size, given that the error bars are fairly large.
Similarly, the desorption probability of OH has been plotted in Fig. 7 as function of
initial OH translational energy in the top three monolayers. Also in this case the desorption probability increases with
increasing initial kinetic energy. The effect is much stronger than for the case of H atoms, which reflects the much
stronger binding energy of OH to its surroundings compared to that of the H atom. Interestingly, if the desorption
probability is weighted with the initial distribution of translational energies, the total OH desorption probability (for the
top three monolayers) increases rapidly at 0.2 eV (the average initial translational energy) and attains a basically
constant value of 0.1% for all energies above that.
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig7.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg30.gif) |
Figure 7:
The probabilities of OH desorption as functions of initial OH translational energy averaged over the
three top monolayers of amorphous ice. Also
shown are the initial distribution of OH translational energies and the total desorption probability
(desorption probability 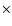 initial distribution). The error bars correspond to a 95% confidence interval.
initial distribution). The error bars correspond to a 95% confidence interval. |
| Open with DEXTER |
The dependence of the indirect H2O desorption probability on the translational energy of the H atom
is found to be weak (see Fig. 8). There is not much evidence of any variation with
translational energy and the desorption probability is fairly constant at 0.1% (averaged over the top six monolayers)
over the whole energy range. It would be natural to expect that there could be a strong dependence on translational
energy, but as discussed above the desorption is in most cases a combined effect of a repulsive force
from the excited molecule, a lowered binding energy, and the momentum transfer from the H atom. Apparently, this
allows also H atoms with relatively low translational energies to kick out H2O molecules.
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig8.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg31.gif) |
Figure 8:
Probability of indirect H2O desorption (``kick-out'') as function of initial H atom
translation energy (divided into bins of 0.5 eV intervals) averaged over the top six monolayers of amorphous ice.
The error bars correspond to a 95% confidence interval. |
| Open with DEXTER |
3.2.4 Dependence on photon energy
Considering the desorption probabilities as functions of the excitation energy (Fig. 9)
it is interesting to note that
H atom desorption becomes less probable with increasing excitation energy (0.8 at 7.3 eV and 0.3 at 9.5 eV).
Since it was shown that the average initial
translational energy mainly increases with increasing excitation energy (Fig. 5)
and that the desorption probability increases with increasing
translational energy (Fig. 6) this seems like a paradox.
The simple explanation of this behavior is that the lower excitation energies
dominate in the top monolayers while the more energetic UV photons are mainly absorbed towards the bulk of the ice
(see, e.g., Fig. 5 of Andersson et al. 2005).
The desorption probability summed over the whole excitation energy range decreases rapidly with depth into the ice
(Table 1) and therefore this unexpected behavior is found. The desorption of OH seems to increase with
increasing excitation energy. This is a reflection of the fact that even though the average initial translational
energy of OH varies only slightly over the excitation energy range (Fig. 5), there is a high-energy tail
of the OH translational energy distribution (see Fig. 7) that becomes larger with higher excitation energies.
The desorption probability of H2O does not show strong dependence on excitation energy,
but considering the rather large error bars some energy dependence cannot be entirely ruled out.
![\begin{figure}
\par\includegraphics[width=7.8cm,clip]{0374fig9.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg32.gif) |
Figure 9:
Probabilities of desorption of H atoms, OH radicals, and H2O molecules
as function of excitation energy averaged over the top six monolayers of amorphous ice.
The error bars correspond to a 95% confidence interval. |
| Open with DEXTER |
3.3 Mobility of photoproducts
The H atoms produced
in the photodissociation event are found to be quite mobile in
the ice. On average the H atoms that become trapped move 8 Å
from their original locations. In extreme cases distances over 70 Å are recorded. The OH radicals formed in the ice move only about 1 Å
with maximum distances moved of 5 Å. However, OH radicals formed from
photodissociation in the top three monolayers are able in some cases
to move tens of Å on top of the surface (up to more than 60 Å). The fact that some of the photofragments are able to move
large distances implies an increased probability of reactions with
other species in or on the ice, than if they would remain in the
immediate vicinity of their point of origin. For more details see
Andersson et al. (2006).
3.4 Comparison to experiments
![\begin{figure}
\par\includegraphics[width=7.8cm,clip]{0374fig10.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/Timg33.gif) |
Figure 10:
Calculated distributions of H atom initial translational energies and desorption energies upon
UV absorption at
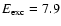 eV summed over the top six monolayers in amorphous ice. eV summed over the top six monolayers in amorphous ice. |
| Open with DEXTER |
In the experiments by Yabushita et al. (2006) on UV irradiation of polycrystalline and
amorphous ices at 100 K absorption at
nm (
eV)
was found to result in H atom desorption.
The translational energy distribution of the desorbing atoms was observed to
consist of three components at 0.61 eV, 0.081 eV, and 0.014 eV, respectively.
The lowest-energy component seems to consist of atoms that have thermalized prior to desorption.
Thermal desorption occurs on a time scale that is likely much longer than would be feasible
to do with molecular dynamics simulations. Therefore, it is not likely that we would see
this third component in our calculations, but the other two should be possible to reproduce.
In Fig. 10 the calculated distributions of
initial translational energies and desorption energies of
the released H atoms following excitation at 7.9 eV are shown. It is seen that the initial translational
energy has a peak at around 1.9 eV and the desorption energy peaks at about the same energy. This clearly is
much higher than found in the experiments, so it seems the energy of the desorbing H atoms is overestimated
in our calculations. There could be three explanations to this behavior: (i) either the H atoms lose more energy
prior to desorption or (ii) they are initially formed with less translational energy or (iii) a combination
of these two effects. The possible sources of loss of highly energetic H atoms in the ice that cannot be treated by
our calculations are: (a) the loss of energy by excitation of intramolecular modes in collisions with H2O molecules
and (b) reactions with H2O molecules to form, e.g., H2 and OH. For a thorough discussion
see Andersson et al. (2006). Since we cannot directly tell how effective mechanism (i) is it
is hard to speculate how much the desorption energy distribution is cooled through energy transfer and reaction.
It is easier to speculate about mechanism (ii), since it is possible to monitor the dependence of the average
desorption energy on the initial translational energy. If the initial translational energy
is around 1 eV the average desorption energies lie in the range 0.4-0.8 eV, which would be in much
better accord with the experimentally measured desorption energies. If this is the most important mechanism for
cooling the desorption energy distribution, then at
eV the initial H atom translational energies
are overestimated by roughly 1 eV. As discussed in Sect. 3.2 this would probably have little importance
for the H2O desorption probability (Fig. 8).
However, the desorption probability of H atoms could be somewhat lower
than predicted in our calculations, but it is still quite likely to be of the same
order of magnitude (Fig. 6). If the average H atom translational energy is overestimated it
is also likely that the OH translational energy is somewhat overestimated. Considering the weak dependence on
excitation energy (Fig. 5) the average OH translational energy might not be highly overestimated,
but the high energy tail could be smaller, meaning that less OH radicals desorb than predicted here.
If the initial kinetic energies of the photofragments are overestimated, the only possibility to account for the
blue shift of the excitation energy is that the intermolecular repulsion is underestimated. This could have as an
interesting effect that the indirect desorption of the surrounding molecules could be underestimated, since
they would experience an even larger repulsive force from the excited molecule than predicted by our calculations.
This remains to be investigated.
The high probabilities of H atom desorption is also in accordance with the
finding of Gerakines et al. (1996) that their
UV-irradiated water ice was most likely depleted of H atoms, since a
large amount of oxygen rich products was found.
Our results on mobility of the released photofragments (Sect. 3.3, Andersson et al. 2006) have been supported by recent
experiments by Elles et al. (2007), who observed
an average separation of H and OH of 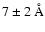 immediately following photodissociation of H2O upon
UV irradiation of liquid water. This is
in excellent agreement with our calculated value of 8
immediately following photodissociation of H2O upon
UV irradiation of liquid water. This is
in excellent agreement with our calculated value of 8 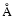 for the average distance from the site of
photodissociation of the H atoms in amorphous ice. Even though the temperatures are quite different
in the two cases, it is expected that liquid water and compact amorphous ice are quite similar on the
short time scales during which photodissociation takes place.
for the average distance from the site of
photodissociation of the H atoms in amorphous ice. Even though the temperatures are quite different
in the two cases, it is expected that liquid water and compact amorphous ice are quite similar on the
short time scales during which photodissociation takes place.
A direct comparison with the water ice photodesorption results by
Westley et al. (1995b,a) is difficult to make
since they used Lyman-
radiation, which leads to excitation to
a higher absorption band than considered here
(Kobayashi 1983). Their findings of basically no photodesorption of intact H2O at
low temperatures in the limit of single-photon absorption can
therefore be neither refuted nor confirmed by our results. However, the detection
of desorbing H2 and O2, and possibly OH and H2O2 actually agree with
the present results (Westley et al. 1995a) (see below).
In the experiments of Öberg et al. (2008) the photodesorbing material
is detected in the form of OH, H2O, H2, and O2. This is the first time
a positive detection of OH photodesorption has been reported. The detection of OH
agrees nicely with our simulations (see also Sect. 4).
It is important to bear in mind that even though
only H, OH, and H2O would desorb upon absorption of a single UV photon,
in the experimental setup a large amount of UV photons may be absorbed in the ice
during a relatively short time interval. This makes it possible to produce
significant amounts of H and OH in the ice, and possibly also O atoms from for
instance the photodissociation of the formed OH radicals. Given that thermal
diffusion is effective in moving these reactive species into close contact,
all the aforementioned desorbing species can be accounted for through recombination
followed by desorption.
4 Discussion and astrophysical implications
Based on the results presented in this paper some important conclusions can be drawn on the possible
outcomes of UV irradiation of water ice in interstellar environments. First, it is important
to realize that by far the most likely species to desorb are H atoms, followed by OH radicals. In addition, when there
is removal of H2O from the surface it seems likely that most of it comes off in the form of separate H and OH fragments.
Therefore, there is not a one-to-one correspondence between H2O molecules removed from a surface and H2O appearing in the gas phase.
So far, this paper has been concerned with probabilities of photoinduced processes following absorption of one UV photon in a specific
layer in the ice. However, not all incident UV photons are absorbed by molecules in the top six monolayers. To estimate
desorption probabilities per incident photon rather than per absorbed photon one needs information
about the absorption cross section. In our semiclassical simulations we are not able to calculate the absolute absorption
cross section. However, Mason et al. (2006) have measured the absorption cross section of water ice at 25 K and found the peak absorption
cross section in the first absorption band to be about
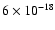 cm2 at an excitation energy of 8.61 eV. This leads to an
absorption probability of 0.007 photons ML-1 (see Appendix A for an outline of this calculation).
cm2 at an excitation energy of 8.61 eV. This leads to an
absorption probability of 0.007 photons ML-1 (see Appendix A for an outline of this calculation).
From the above estimate of the absorption probability it is possible to calculate photodesorption probabilities per incident UV photon.
This is done by weighting the desorption probabilities per absorbed photon for each monolayer by the absorption probability
for the specific monolayer with the absorption probabilities in any upper monolayers subtracted from the incoming photon flux.
Using the information in Table 2 one arrives at a probability of removal of H2O from the ice of
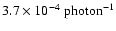 .
About 60% of the removed H2O comes off in the form of H + OH, 20% desorbs as recombined
H2O, and the remaining 20% consists of H2O ``kicked'' out from the surface. The total photodesorption yield of intact H2O
molecules is
.
About 60% of the removed H2O comes off in the form of H + OH, 20% desorbs as recombined
H2O, and the remaining 20% consists of H2O ``kicked'' out from the surface. The total photodesorption yield of intact H2O
molecules is
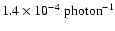 .
For OH desorption the probability is
.
For OH desorption the probability is
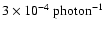 ,
which includes both desorption with and without H atoms. The H atom photodesorption probability
is relatively high,
,
which includes both desorption with and without H atoms. The H atom photodesorption probability
is relatively high,
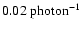 .
The above ratios of photodesorption of OH and H2O are in excellent agreement with the recent experiments by Öberg et al. (submitted to ApJ),
which inferred that roughly equal amounts of OH and H2O photodesorb at low surface temperature (18 K).
They obtain a total photodesorption yield of about
.
The above ratios of photodesorption of OH and H2O are in excellent agreement with the recent experiments by Öberg et al. (submitted to ApJ),
which inferred that roughly equal amounts of OH and H2O photodesorb at low surface temperature (18 K).
They obtain a total photodesorption yield of about
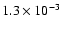 in the low-temperature limit, which is about 3 times higher
than what is found from our simulations. Given the experimental uncertainties and the approximations made in the simulations this
can be considered as good agreement.
Commonly used estimates of H2O
photodesorption probabilities in the range
in the low-temperature limit, which is about 3 times higher
than what is found from our simulations. Given the experimental uncertainties and the approximations made in the simulations this
can be considered as good agreement.
Commonly used estimates of H2O
photodesorption probabilities in the range
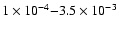 have been used to model different environments in
agreement with observations (Snell et al. 2005; Bergin et al. 1995; Willacy & Langer 2000; Bergin & Melnick 2005; Dominik et al. 2005). Our results indicate that these estimates are reasonable.
However, the finding that less than half of the desorbed material leaves the grain in the form of intact H2O molecules
is something that should be included in the models.
have been used to model different environments in
agreement with observations (Snell et al. 2005; Bergin et al. 1995; Willacy & Langer 2000; Bergin & Melnick 2005; Dominik et al. 2005). Our results indicate that these estimates are reasonable.
However, the finding that less than half of the desorbed material leaves the grain in the form of intact H2O molecules
is something that should be included in the models.
The results presented here are only for photoinduced processes that are followed until the photoproducts desorb or are thermalized within the
ice. This is all happening on a picosecond time scale. For longer time scales there is the possibility of thermal desorption of especially
the H atoms. These are relatively weakly bound to the ice surface and it is quite probable that some fraction of the released H atoms
desorb after thermalization in the ice. This contribution to the desorption probability is therefore not included in the above estimate.
It is interesting to discuss the possible effects of the overall morphology of the ice surface.
It is clear that the vast majority of water ice surfaces in the interstellar medium are amorphous (Hagen et al. 1981). However, it is debated
whether the ice is mostly porous, as found in vapor deposited ice (Mayer & Pletzer 1986; Kimmel et al. 2001b,a), or more compact (Fraser et al. 2004; Palumbo 2006).
A complicating factor is that most likely the ice is formed through chemical reactions on grains rather than accretion of H2O
molecules from the gas phase (O'Neill & Williams 1999). Therefore, the exact ice morphology that results from such a chemical build-up of the
ice is not yet clear (see however Cuppen & Herbst 2007). If a porous ice surface is subjected to UV irradiation one could have release of H, OH, or H2O into a void
in the ice rather than directly into the gas phase. This is inferred to happen in the experiments by Yabushita et al. (2006) where a large
component of the desorbing H atoms released after UV irradiaton of amorphous ice are thermalized, likely due to H atoms being accommodated
within a void in the ice and then desorbing thermally. Some OH and H2O could photodesorb in a similar way. However, if the
path to reach the gas phase from inside a pore is restricted, these species have a high probability of being trapped inside
a pore because of their strong attractive interactions with the ice. In practice, the OH and H2O released in this
way might not show up in desorption. Therefore, if a porous ice is considered it is necessary to distinguish
between the surface area that is directly exposed to the gas phase and that which is within a void with restricted access to the outside.
In conclusion, the H atom desorption from a porous ice is likely to be different from that of compact non-porous ice, but
the desorption yields of OH and H2O are not necessarily very different in the two types of ice.
The cosmic ray induced UV flux inside dense clouds is about
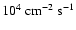 (Shen et al. 2004).
For an ice-coated grain with a typical
size of 0.1
(Shen et al. 2004).
For an ice-coated grain with a typical
size of 0.1 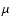 m, this would give an arrival rate of about 1 UV photon per day. The case of a single UV absorption
event as described in our simulations therefore gives a realistic picture of photodesorption in dense clouds. In the laboratory, the UV flux is many orders of magnitude higher and multiple absorptions of UV photons within the ice surface in a short time interval may
drive secondary reactions of photodissociation products from different H2O molecules. Experiments with different UV flux levels
down to low levels will be needed to provide quantitative data on photodesorption yields relevant for astrophysical applications.
m, this would give an arrival rate of about 1 UV photon per day. The case of a single UV absorption
event as described in our simulations therefore gives a realistic picture of photodesorption in dense clouds. In the laboratory, the UV flux is many orders of magnitude higher and multiple absorptions of UV photons within the ice surface in a short time interval may
drive secondary reactions of photodissociation products from different H2O molecules. Experiments with different UV flux levels
down to low levels will be needed to provide quantitative data on photodesorption yields relevant for astrophysical applications.
As has also been noted in our previous work another important aspect is the release of reactive species into the ice. That would
have implications for reactivity in the ice with, e.g., CO that could react with energetic H or OH to form HCO or CO2. This could be
one clue to unraveling the mystery of CH3OH and CO2 formation in the interstellar medium. Indeed, formation of CO2 is readily observed
when a mixed H2O:CO ice is photolysed (Watanabe et al. 2007; d'Hendecourt et al. 1986; Watanabe & Kouchi 2002).
We have shown that it is possible to have H2O photodesorption upon UV absorption to the first absorption band
in the top five monolayers of an amorphous ice surface.
The main mechanisms for this photodesorption are either photodissociation followed by recombination of H and OH and subsequent desorption
of the recombined H2O molecule or a ``kick-out'' of another H2O molecule in the ice by the energetic H atom released from photodissociation
or, less likely, by the energy released from a recombined H2O molecule. In most cases, however, removal of an H2O molecule from the ice is
in the form of separate H and OH fragments. An estimate of the photodesorption yield per incident UV photon
from our calculations agrees well with the H2O photodesorption yields that are commonly used in modeling astrophysical environments.
UV absorption leads in most cases to desorption of H atoms or the trapping of H and OH in the ice either as separate fragments or as recombined H2O.
The desorption of H atoms is about 2 or 3 orders of magnitude more probable than desorption of OH and H2O. The OH desorption probability is about twice the H2O
desorption probability. The high mobility of H atoms inside the ice and OH radicals on the ice surface will facilitate formation of other
molecules such as CO2.
Acknowledgements
We thank Karin Öberg, Herma Cuppen, and Geert-Jan Kroes for stimulating discussions.
Some of the calculations reported here were performed at Chalmers Centre for Computational
Science and Engineering (C3SE) computing resources. This research was funded by a Netherlands Organization for Scientific Research (NWO) Spinoza
grant (for one of the authors (E.F.v.D)) and a NWO-CW Top grant.
Appendix A: Estimate of absorption probability
To calculate an absorption probability per monolayer in an interstellar ice surface one needs
an estimate of the effective area taken up by one molecule. Since the angle of incidence of the photon is arbitrary,
all possible incidence angles have to be taken into account, not only normal incidence.
The surface area of our simulation cell is
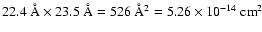 .
If one assumes a flat surface, the average effective surface area seen by a photon from any incidence angle between 0
.
If one assumes a flat surface, the average effective surface area seen by a photon from any incidence angle between 0 (normal incidence)
and 90
(parallel to the surface) is the actual surface area divided by two. This is arrived upon by
the following expression:
(normal incidence)
and 90
(parallel to the surface) is the actual surface area divided by two. This is arrived upon by
the following expression:
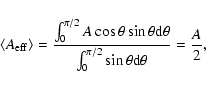 |
(A.1) |
with the effective area given by
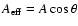 where A is the actual surface area and
where A is the actual surface area and 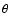 is the angle of incidence. The average effective surface area of the simulation cell is
is the angle of incidence. The average effective surface area of the simulation cell is
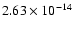 cm2.
Each monolayer consists of 30 H2O molecules. A single molecule will therefore have an average effective area
cm2.
Each monolayer consists of 30 H2O molecules. A single molecule will therefore have an average effective area
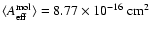 .
Mason et al. (2006) measured the peak absorption cross section,
.
Mason et al. (2006) measured the peak absorption cross section, 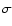 ,
in the first absorption band to be about
cm2around an excitation energy of 8.61 eV.
With this value the absorption probability per monolayer becomes
,
in the first absorption band to be about
cm2around an excitation energy of 8.61 eV.
With this value the absorption probability per monolayer becomes
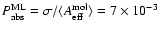 (this number may vary somewhat with
excitation energy). If one
considers an infinitely deep ice surface all photons will be absorbed within the ice.
For an ice surface in the interstellar medium consisting of a finite number of layers,
the value of
(this number may vary somewhat with
excitation energy). If one
considers an infinitely deep ice surface all photons will be absorbed within the ice.
For an ice surface in the interstellar medium consisting of a finite number of layers,
the value of
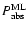 may vary depending on the shape of the grain,
since the surface is not necessarily flat. However, it is likely that any photon that passes through an ice mantle will be
absorbed by the silicate grain, leading to almost total absorption of the incident photons within the grain.
may vary depending on the shape of the grain,
since the surface is not necessarily flat. However, it is likely that any photon that passes through an ice mantle will be
absorbed by the silicate grain, leading to almost total absorption of the incident photons within the grain.
- Aikawa, Y., van
Zadelhoff, G. J., van Dishoeck, E. F., & Herbst, E. 2002,
A&A, 386, 622 [NASA ADS] [CrossRef] [EDP Sciences]
- Al-Halabi, A., van
Dishoeck, E. F., & Kroes, G. J. 2004a,
J. Chem. Phys., 120, 3358 [NASA ADS] [CrossRef]
(In the text)
- Al-Halabi, A., Fraser,
H. F., Kroes, G. J., & van Dishoeck, E. F. 2004b, A&A, 422,
777 [NASA ADS] [CrossRef] [EDP Sciences]
- Allen, M. P., &
Tildesley, D. J. 1987, Computer Simulations of Liquids (Oxford:
Clarendon)
(In the text)
- Andersson, S., Kroes, G.
J., & van Dishoeck, E. F. 2005, Chem. Phys. Lett., 408,
415 [NASA ADS] [CrossRef]
(In the text)
- Andersson, S., Al-Halabi,
A., Kroes, G. J., & van Dishoeck, E. F. 2006,
J. Chem. Phys., 124, 064715 [NASA ADS] [CrossRef]
(In the text)
- Berendsen, H. J. C.,
Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J.
R. 1984, J. Chem. Phys., 81, 3684 [NASA ADS] [CrossRef]
(In the text)
- Bergeld, J., &
Chakarov, D. 2006, J. Chem. Phys., 125, 141103 [NASA ADS] [CrossRef]
- Bergin, E. A., &
Melnick, G. 2005, in Astrochemistry: recent successes and current
challenges, ed. D. C. Lis, G. A. Blake, & E. Herbst (Cambridge:
Cambridge University Press), IAU Symp., 231, 309
(In the text)
- Bergin, E. A., Langer, W.
D., & Goldsmith, P. F. 1995, ApJ, 441, 222 [NASA ADS] [CrossRef]
- Bergin, E. A., Alves, J.,
Huard, T., & Lada, C. J. 2002, ApJ, 570, L101 [NASA ADS] [CrossRef]
- Boonman, A. M. S., Doty,
S. D., van Dishoeck, E. F., et al. 2003, A&A, 406,
937 [NASA ADS] [CrossRef] [EDP Sciences]
- Caselli, P., Walmsley, C.
M., Tafalla, M., Dore, L., & Myers, P. C. 1999, ApJ, 523,
L165 [NASA ADS] [CrossRef]
- Ceccarelli, C., &
Dominik, C. 2005, A&A, 440, 583 [NASA ADS] [CrossRef] [EDP Sciences]
- Cuppen, H. M., &
Herbst, E. 2007, ApJ, 668, 294 [NASA ADS] [CrossRef]
(In the text)
- Dalgarno, A., Yan, M.,
& Liu, W. 1999, ApJS, 125, 237 [NASA ADS] [CrossRef]
- d'Hendecourt, L. B.,
Allamandola, L. J., Baas, F., & Greenberg, J. M. 1982, A&A,
109, L12 [NASA ADS]
- d'Hendecourt, L. B.,
Allamandola, L. J., Grim, R. J. A., & Greenberg, J. M. 1986,
A&A, 158, 119 [NASA ADS]
- Dobbyn, A. J., &
Knowles, P. J. 1997, Mol. Phys., 91, 1107 [NASA ADS] [CrossRef]
(In the text)
- Dominik, C., Ceccarelli,
C., Hollenbach, D., & Kaufman, M. 2005, ApJ, 635, L85 [NASA ADS] [CrossRef]
- Ehrenfreund, P.,
d'Hendecourt, L., Charnley, S., & Ruiterkamp, R. 2001, J.
Geophys. Res., 106, 33291 [NASA ADS] [CrossRef]
- Elles, C. G., Shkrob, I.
A., Crowell, R. A., & Bradforth, S. E. 2007,
J. Chem. Phys., 126, 164503 [NASA ADS] [CrossRef]
(In the text)
- Fraser, H. J., Collings,
M. P., Dever, J. W., & McCoustra, M. R. S. 2004, MNRAS, 353,
59 [NASA ADS] [CrossRef]
- Garrod, R. T., &
Herbst, E. 2006, A&A, 457, 927 [NASA ADS] [CrossRef] [EDP Sciences]
- Gerakines, P. A.,
Schutte, W. A., & Ehrenfreund, P. 1996, A&A, 312, 289 [NASA ADS]
(In the text)
- Ghormley, J. A., &
Hochanadel, C. J. 1971, J. Phys. Chem., 75, 40 [CrossRef]
(In the text)
- Gibb, E. L., Whittet, D.
C. B., Schutte, W. A., et al. 2000, ApJ, 536, 347 [NASA ADS] [CrossRef]
- Hagen, W., Tielens, A. G.
G. M., & Greenberg, J. M. 1981, Chem. Phys., 56, 367 [CrossRef]
(In the text)
- Hartquist, T. W., &
Williams, D. A. 1990, MNRAS, 247, 343 [NASA ADS]
- Huber, K. P., &
Herzberg, G. 1979, Molecular Spectra and Molecular Structure, IV,
Constants of Diatomic Molecules (New York: Van Nostrand)
(In the text)
- Jewitt, D. C., & Luu,
J. 2004, Nature, 432, 731 [NASA ADS] [CrossRef]
(In the text)
- Jorgensen, W. L.,
Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L.
1983, J. Chem. Phys., 79, 926 [NASA ADS] [CrossRef]
(In the text)
- Kimmel, G. A.,
Stevenson, K. P., Dohnálek, Z., Scott Smith, R., & Kay, B.
D. 2001a, J. Chem. Phys., 114, 5284 [NASA ADS] [CrossRef]
- Kimmel, G. A.,
Dohnálek, Z., Stevenson, K. P., Scott Smith, R., & Kay, B.
D. 2001b, J. Chem. Phys., 114, 5295 [NASA ADS] [CrossRef]
- Kobayashi, K. 1983, J.
Phys. Chem., 87, 4317 [CrossRef]
(In the text)
- Léger, A., Jura, M.,
& Omont, A. 1985, A&A, 144, 147 [NASA ADS]
- Mason, N. J., Dawes, A,
Holtom, P. D., et al. 2006, Faraday Discuss., 133, 311 [CrossRef]
(In the text)
- Mayer, E., & Pletzer,
R. 1986, Nature, 319, 298 [NASA ADS] [CrossRef]
- Mumma, M. J., Weissman,
P. R., & Stern, S. A. 1993, in Protostars &
Planets III, ed. E. H. Levy, & J. I. Lunine, Tucson,
University of Arizona, 1177
(In the text)
- Murakawa, K., Tamura, M.,
& Nagata, T. 2000, ApJS, 128, 603 [NASA ADS] [CrossRef]
- Nishi, N., Shinohara, H.,
& Okuyama, T. 1984, J. Chem. Phys., 80, 3898 [NASA ADS] [CrossRef]
- O'Neill, P. T., &
Williams, D. A. 1999, Ap&SS, 266, 539 [NASA ADS] [CrossRef]
(In the text)
- Öberg, K. I., Visser, R., van
Dishoeck, E. F., & Linnartz, H. 2008, ApJ, submitted
- Palumbo, M. E. 2006,
A&A, 453, 903 [NASA ADS] [CrossRef] [EDP Sciences]
- Pontoppidan, K. M. 2006,
A&A, 453, L47 [NASA ADS] [CrossRef] [EDP Sciences]
- Pontoppidan, K. M., van
Dishoeck, E. F., & Dartois, E. 2004, A&A, 426, 925 [NASA ADS] [CrossRef] [EDP Sciences]
- Pontoppidan, K. M.,
Dullemond, C. P., van Dishoeck, E. F., et al. 2005, ApJ, 622,
463 [NASA ADS] [CrossRef]
- Prasad, S. S., &
Tarafdar, S. P. 1983, ApJ, 267, 603 [NASA ADS] [CrossRef]
- Schinke, R. 1993,
Photodissociation Dynamics (Cambridge: Cambridge University
Press)
(In the text)
- Shen, C., Greenberg, J.
M., Schutte, W. A., & van Dishoeck, E. F. 2004, A&A, 415,
203 [NASA ADS] [CrossRef] [EDP Sciences]
- Smith, R. G., Sellgren,
K., & Tokunaga, A. T. 1989, ApJ, 344, 413 [NASA ADS] [CrossRef]
- Snell, R. L., Howe, J.
E., Ashby, M. L. N., et al. 2000, ApJ, 539, L97 [NASA ADS] [CrossRef]
- Snell, R. L., Hollenbach,
D., Howe, J. E., et al. 2005, ApJ, 620, 758 [NASA ADS] [CrossRef]
- Spaans, M., Hogerheijde,
M. R., Mundy, L. G., & van Dishoeck, E. F. 1995, ApJ, 455,
L167 [NASA ADS]
(In the text)
- Stäuber, P., Doty,
S. D., van Dishoeck, E. F., Jørgensen, J. K., & Benz, A.
O. 2004, A&A, 425, 577 [NASA ADS] [CrossRef] [EDP Sciences]
- Stäuber, P., Doty,
S. D., van Dishoeck, E. F., & Benz, A. O. 2005, A&A, 440,
949 [NASA ADS] [CrossRef] [EDP Sciences]
- Terada, H., Tokunaga, A.
T., Kobayashi, et al. 2007, ApJ, 667, 303 [NASA ADS] [CrossRef]
- van Dishoeck, E. F.,
Helmich, F. P., de Graauw, T., et al. 1996, A&A, 315,
L349 [NASA ADS]
- van Harrevelt, R.,
& van Hemert, M. C. 2000, J. Chem. Phys., 112,
5777 [NASA ADS] [CrossRef]
(In the text)
- van Harrevelt, R., van
Hemert, M. C. & Schatz, G. C. 2001, J. Phys. Chem. A, 105,
11480 [CrossRef]
(In the text)
- Watanabe, N., &
Kouchi, A. 2002, ApJ, 567, 651 [NASA ADS] [CrossRef]
- Watanabe, N., Horii, T.,
& Kouchi, A. 2000, ApJ, 541, 772 [NASA ADS] [CrossRef]
(In the text)
- Watanabe, N., Mouri, O.,
Nagaoka, A., et al. 2007, ApJ, 668, 1001 [NASA ADS] [CrossRef]
- Westley, M. S.,
Baragiola, R. A., Johnson, R. E., & Baratta, G. A. 1995a,
Planet. Space Sci., 43, 1311 [NASA ADS] [CrossRef]
- Westley, M. S.,
Baragiola, R. A., Johnson, R. E., & Baratta, G. A. 1995b,
Nature, 373, 405 [NASA ADS] [CrossRef]
- Whittet, D. C. B., Bode,
M. F., Longmore, A. J., et al. 1988, MNRAS, 233, 321 [NASA ADS]
- Willacy, K., &
Langer, W. D. 2000, ApJ, 544, 903 [NASA ADS] [CrossRef]
- Willner, S. P., Gillett,
F. C., Herter, T. L., et al. 1982, ApJ, 253, 174 [NASA ADS] [CrossRef]
- Yabushita, A., Kanda, D.,
Kawanaka, N., Kawasaki, M., & Ashfold, M. N. R. 2006,
J. Chem. Phys., 125, 133406 [NASA ADS] [CrossRef]
(In the text)
Copyright ESO 2008
![\begin{figure}
\par\includegraphics[angle=270,width=8cm,clip]{0374fig1.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img12.gif)
![\begin{figure}
\par\includegraphics[angle=270,width=8cm,clip]{0374fig2.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img15.gif)
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig3.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img21.gif)
![\begin{figure}
\par\includegraphics[angle=270,width=8.4cm,clip]{0374fig4.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/img24.gif)
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig5.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img25.gif)
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig6.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img29.gif)
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig7.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/img30.gif)
![\begin{figure}
\par\includegraphics[width=8cm,clip]{0374fig8.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/img31.gif)
![\begin{figure}
\par\includegraphics[width=7.8cm,clip]{0374fig9.eps}
\end{figure}](/articles/aa/full/2008/45/aa10374-08/img32.gif)
![\begin{figure}
\par\includegraphics[width=7.8cm,clip]{0374fig10.eps} \end{figure}](/articles/aa/full/2008/45/aa10374-08/img33.gif)