the reaction C
A&A 376, 663-666 (2001)
DOI: 10.1051/0004-6361:20010998
D. Talbi1 - E. Herbst2
1 - LETMEX, Université de Nice Sophia Antipolis, Parc Valrose, UFR Sciences, 06108 Nice Cedex 2, France
2 -
Departments of Physics and Astronomy, The Ohio State University, Columbus, OH 43210, USA
Received 16 March 2001 / Accepted 6 July 2001
Abstract
Although deuterium isotope fractionation in cold, dark
interstellar clouds is reasonably well understood via gas-phase
chemistry, there are some
discrepancies between observation and theory. For example, the
observed
abundance ratio between the deuterated species C3HD and the
cyclic molecule C3H2 is significantly higher than most
theoretical values unless the exchange reaction
C3H3++ HD
![]() C3H2D++H2 is efficient. In this paper, we report quantum chemical and
dynamical calculations on this reaction, which show it to possess a
large activation energy barrier and to be very slow at all normal
interstellar temperatures.
C3H2D++H2 is efficient. In this paper, we report quantum chemical and
dynamical calculations on this reaction, which show it to possess a
large activation energy barrier and to be very slow at all normal
interstellar temperatures.
Key words: ISM: abundances - ISM: molecules - molecular processes
Deuterium isotopic fractionation is a continuing topic of interest in
the study of dense interstellar clouds. Although the deuterium to hydrogen
elemental abundance ratio is only
![]() (e.g.
Wright et al. 1999), the abundance ratios of singly deuterated
trace isotopomers such as H2D+, DCO+, NH2D, DCN, etc.
to their respective normal species are measured to be 0.01-0.10 or even
higher (depending on excitation model) in such
clouds. Indeed, the doubly deuterated isotopomer NHD2 has
recently been detected in the dark cloud L134N with an abundance ratio
NHD2/NH2D of
(e.g.
Wright et al. 1999), the abundance ratios of singly deuterated
trace isotopomers such as H2D+, DCO+, NH2D, DCN, etc.
to their respective normal species are measured to be 0.01-0.10 or even
higher (depending on excitation model) in such
clouds. Indeed, the doubly deuterated isotopomer NHD2 has
recently been detected in the dark cloud L134N with an abundance ratio
NHD2/NH2D of
![]() (Roueff et al. 2000), which is
similar to the value measured for NH2D/NH3 of
(Roueff et al. 2000), which is
similar to the value measured for NH2D/NH3 of
![]() (Tiné et al. 2000).
(Tiné et al. 2000).
It is well known that for cold dark clouds, exothermic exchange
reactions between the abundant ions H3+, CH3+ and
C2H2+ and HD are very important in producing enhanced
abundances of deuterated ions, which then undergo further reactions to
produce other deuterium-containing species. Models for dark clouds
including these and other reactions have been reported by Millar
et al. (1989) and more recently by Roberts & Millar
(2000a,2000b) and by Turner (2001). In these models,
gas-phase reactions generally can account for the abundances of singly
and doubly deuterated isotopomers, although the agreement is not
uniform. One of the problems appears to be the large
deuterium fractionation involving the cyclic molecule C3H2.
Early measurements by Bell et al. (1988) obtained that
C3HD/C3H
![]() depending upon position
in TMC-1. More recent measurements by Turner (2001) in TMC-1
show that the ratio appears to be closer to 0.048. Calculated values
tend to be on the order of
depending upon position
in TMC-1. More recent measurements by Turner (2001) in TMC-1
show that the ratio appears to be closer to 0.048. Calculated values
tend to be on the order of
![]() (Millar et al.
1989; Roberts & Millar 2000a; Turner 2001)
unless there is significant freeze out of molecules onto dust (Roberts
& Millar 2000a,2000b), in which case all abundance ratios of
deuterated to normal isotopomers increase. To complicate matters
though, a somewhat higher value of 0.032 has been calculated by Turner
(2001) for TMC-1 at early time.
(Millar et al.
1989; Roberts & Millar 2000a; Turner 2001)
unless there is significant freeze out of molecules onto dust (Roberts
& Millar 2000a,2000b), in which case all abundance ratios of
deuterated to normal isotopomers increase. To complicate matters
though, a somewhat higher value of 0.032 has been calculated by Turner
(2001) for TMC-1 at early time.
In 1993, Howe & Millar (1993) suggested that if the exothermic
left-to-right reaction of the reaction system
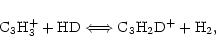 |
(1) |
An extensive investigation of the singlet potential energy surface
corresponding to an HD molecule approaching the c-C3H3+ion was initially undertaken at the RHF and MP2 levels in combination
with a triple split-valence basis set to which diffuse orbitals have
been added together with polarization functions (namely
6-311+G(2d,p)). The stationary points located on this surface were
reoptimized using the density functional formalism (B3LYP functional;
Becke 1993) and their character (either minimum energy point,
for which all vibrational frequencies are real, or saddle point,
characterized by one imaginary frequency) was confirmed by
vibrational analysis done at the same B3LYP level. For accurate
electronic energies, single point calculations were performed at the
CCSD(T)/6-311+G(3df,2pd) theoretical level using the
B3LYP/6-311+G(2d,p) optimized geometries (hereafter
CCSD(T)/6-311+G(3df,2pd)//B3LYP/6-311+G(2d,p)). This is a coupled
cluster singles and doubles method with a perturbative treatment of
the triple excitations (Raghavachari et al. 1989), employing
the previous triple-zeta basis set to which extra sets of polarization
functions have been added. With such a basis set we reach a
flexibility in the single determinant representation that approaches
the Hartree-Fock limit. All energies were corrected for zero-point
energy (ZPE) contributions calculated at the B3LYP/6-311+G(2d,p)
level and scaled by a factor of 0.9613 (Frisch et al. 1996).
When relative energies were small (for the case of the long-distance
complexes in the entrance and exit channels) they were adjusted for
the basis set superposition error (BSSE) following the counterpoise
procedure (Boys & Bernardi 1970; Liu & McLean 1973,
1989). All the quantum chemical
calculations of this study were performed by means of the Gaussian 98
package (Frisch et al. 1998). Uncertainties in calculated
energies at the highest level of calculation are estimated to be 1-2
kcal/mol.
![\begin{figure}
\par\includegraphics[width=10cm]{ms1283f1.eps}\end{figure}](/articles/aa/full/2001/35/aa1283/img12.gif) |
Figure 1:
CCSD(T)/6-311+G(3df,2pd//B3LYP/6-311+G(2d,p) lowest energy
reaction pathway for the reaction system
C3H3+ + HD
|
| Open with DEXTER | |
![\begin{figure}
\par\includegraphics[width=8.8cm]{ms1283f2.eps}\end{figure}](/articles/aa/full/2001/35/aa1283/img13.gif) |
Figure 2: Structures of the (C3H4D+) stationary points optimized at the B3LYP/6-311+G(2d,p) level. The distances are in Å. |
| Open with DEXTER | |
The CCSD(T)/6-311+G(3df,2pd)//B3LYP /6-311+ G(2d,p) energy profile of
the c-C3H3+ + HD reaction is shown in Fig. 1 and the
structures of the corresponding (C3H4D+) stationary
points, optimized at the B3LYP/6-311+G(2d,p) level, are given in
Fig. 2. The energy profile reveals the existence on the reaction
path of a high transition state, labelled (C3H2...
H2D+)![]() in Fig. 2, which can be described as a complex
between c-C3H2 and H2D+.
This transition state corresponds to the
minimum energy needed to go from reactants to products and is best
thought of as a saddle point in the potential barrier. Its
vibrational analysis shows one imaginary frequency of 1658 cm-1 corresponding to a stretching of the two top hydrogens toward
dissociation. The shallow minimum in the entrance channel
corresponds to a long-distance complex between c-C3H3+ and HD (see Fig. 2). Its binding energy after ZPE and BSSE
corrections are made is -0.4 kcal/mol, which is on the order of the
uncertainty in our calculations. The presence of a minimum energy
structure on this part of the potential energy surface is therefore
not proved and needs more investigation. However, whether this loose
complex exists or not is of no importance for the present study
considering the height (
in Fig. 2, which can be described as a complex
between c-C3H2 and H2D+.
This transition state corresponds to the
minimum energy needed to go from reactants to products and is best
thought of as a saddle point in the potential barrier. Its
vibrational analysis shows one imaginary frequency of 1658 cm-1 corresponding to a stretching of the two top hydrogens toward
dissociation. The shallow minimum in the entrance channel
corresponds to a long-distance complex between c-C3H3+ and HD (see Fig. 2). Its binding energy after ZPE and BSSE
corrections are made is -0.4 kcal/mol, which is on the order of the
uncertainty in our calculations. The presence of a minimum energy
structure on this part of the potential energy surface is therefore
not proved and needs more investigation. However, whether this loose
complex exists or not is of no importance for the present study
considering the height (
![]() kcal/mol above the reactants) of the
following transition state barrier. This argument is equally valid
for the loose (C3H2D+-H2) complex in the exit
channel, which corresponds to another shallow minimum in the
potential energy profile. Table 1 contains vibrational frequencies
and rotational constants for the three intermediate stationary points.
kcal/mol above the reactants) of the
following transition state barrier. This argument is equally valid
for the loose (C3H2D+-H2) complex in the exit
channel, which corresponds to another shallow minimum in the
potential energy profile. Table 1 contains vibrational frequencies
and rotational constants for the three intermediate stationary points.
| Species | Harmonic frequencies a | Rotational constants b |
| (cm-1) | (GHz) | |
| C3H
|
|
30.26996 |
| 1016, 1259, 1261, 1590, 3082 | 8.00741 | |
| 3113, 3151, 3653 | 6.33231 | |
|
|
||
| (C3H
|
|
30.88821 |
| 1110, 1289, 1406, 1588, 2379 | 11.11820 | |
| 2930, 3115, 3147 | 8.17545 | |
|
|
||
| (C3H2D
|
|
30.43985 |
| 984, 1238, 1252, 1546, 2329 | 9.90485 | |
| 3113, 3144, 4217 | 7.47315 | |
|
|
The rate coefficient for the reaction between C3H3+ and
HD (or the reverse reaction) can be calculated with "activated complex''
theory (Weston
& Schwarz 1972). In this method, an equilibrium is assumed to occur
between the reactants and the transition state (see Fig. 1), while the
weakly-bound entrance and exit channels complexes are ignored.
Activated complex theory does not give us the probability that the
transition state decays to products rather than reactants, but at low
energies, the exothermic channel is preferred.
With the assumption of decay into exothermic products, the rate
coefficient for the left-to-right reaction in system (1) is given by the equation
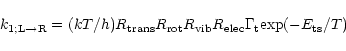 |
(2) |
The dominant term in this expression is the exponential
one. From Fig. 1, it can be seen that
![]() kcal/mol
kcal/mol
![]() K. Thus even at temperatures approaching
room temperature (300 K), the very large negative value of the
exponent (
K. Thus even at temperatures approaching
room temperature (300 K), the very large negative value of the
exponent (
![]() )
renders the rate coefficient effectively
zero whatever
the values of the other terms. Nevertheless, for completeness we have
evaluated the other factors in Eq. (2) (Herbst & Talbi 1998).
At 10 K we obtain
)
renders the rate coefficient effectively
zero whatever
the values of the other terms. Nevertheless, for completeness we have
evaluated the other factors in Eq. (2) (Herbst & Talbi 1998).
At 10 K we obtain
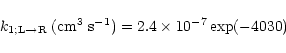 |
(3) |
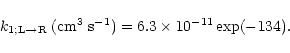 |
(4) |
Acknowledgements
The Astrochemistry Program at The Ohio State University is supported by The National Science Foundation. Parts of the calculations reported here were supported by the IDRIS (France); the support is gratefully acknowledged.